As filed with the Securities and Exchange Commission on February 10, 2020
Registration No. 333‑
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM S‑3
REGISTRATION STATEMENT UNDER THE SECURITIES ACT OF 1933
BRICKELL BIOTECH, INC.
(Exact name of registrant as specified in its charter)
Delaware (State or other jurisdiction of incorporation or organization) | 93-0948554 (I.R.S. Employer Identification No.) |
5777 Central Avenue
Suite 102
Boulder, CO 80301
(720) 505-4755
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
Robert B. Brown
Chief Executive Officer
5777 Central Avenue
Suite 102
Boulder, CO 80301
(720) 505-4755
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Please send copies of all communications to: |
Anna T. Pinedo, Esq. Brian D. Hirshberg, Esq. Mayer Brown LLP 1221 Avenue of the Americas New York, NY 10020 Telephone: (212) 506‑2500 |
Approximate date of commencement of proposed sale to the public:
From time to time after the effective date of this registration statement.
If the only securities being registered on this Form are being offered pursuant to dividend or interest reinvestment plans, please check the following box: o
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, other than securities offered only in connection with dividend or interest reinvestment plans, check the following box: x
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering: o
If this Form is a post‑effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering: o
If this Form is a registration statement pursuant to General Instruction I.D. or a post‑effective amendment thereto that shall become effective upon filing with the Commission pursuant to Rule 462(e) under the Securities Act, check the following box: o
If this Form is a post‑effective amendment to a registration statement filed pursuant to General Instruction I.D. filed to register additional securities or additional classes of securities pursuant to Rule 413(b) under the Securities Act, check the following box: o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer o | Accelerated filer o |
Non-accelerated filer o | Smaller reporting company x |
Emerging growth company o | |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 7(a)(2)(B) of Securities Act. o
CALCULATION OF REGISTRATION FEE
Title of each class of Securities to be registered | Amount to be registered (1) | Proposed maximum offering price per unit (2) | Proposed maximum aggregate offering price (2) | Amount of registration fee | ||||||
Common stock, par value $0.01 per share(3) | ||||||||||
Preferred stock, par value $0.01 per share(3) | ||||||||||
Debt securities(3) | ||||||||||
Warrants(3) | ||||||||||
Units(3) | ||||||||||
Total: | $75,000,000(4) | $75,000,000 | $9,735 | |||||||
(1) | There are being registered hereunder such indeterminate number of common stock and preferred stock, debt securities, warrants and/or units of the registrant as shall have an aggregate initial offering price not to exceed $75,000,000. Any securities registered hereunder may be sold separately or in combination with other securities registered hereunder. The proposed maximum offering price of the securities will be determined, from time to time, by the registrant in connection with the issuance by the registrant of the securities registered hereunder. The securities registered also include such indeterminate amounts and numbers of common stock as may be issued upon conversion of or exchange for preferred stock, debt securities, warrants or units that provide for such conversion or exchange. The amount of each class of securities being registered under this registration statement is not specified pursuant to General Instruction II.D. of Form S-3 under the Securities Act of 1933, as amended (the “Securities Act”). | |||||||||
(2) | The proposed maximum aggregate offering price has been estimated solely for the purpose of calculating the registration fee pursuant to Rule 457(o) under the Securities Act. A filing fee of $4,755 is being paid in connection with the filing of this registration statement. Pursuant to Rule 415(a)(6) under the Securities Act, the securities registered hereunder include $40,000,000 in aggregate offering price of unsold securities (“Unsold Securities”) previously registered pursuant to a registration statement on Form S-3 (File No. 333-225208) originally filed with the Securities and Exchange Commission by Vical Incorporated, the registrant’s predecessor, on May 25, 2018 and declared effective on June 11, 2018 (the “Prior Registration Statement”). Pursuant to Rule 415(a)(6) under the Securities Act, the filing fee of $4,980 relating to the Unsold Securities under the Prior Registration Statement, which was paid under the Prior Registration Statement, will continue to be applied to the Unsold Securities registered pursuant to this registration statement. Pursuant to Rule 415(a)(6), the offering of the Unsold Securities under the Prior Registration Statement will be deemed terminated as of the date of effectiveness of this registration statement. | |||||||||
(3) | Pursuant to Rule 416 under the Securities Act, an indeterminate number of additional securities are registered hereunder that may be issued to prevent dilution in connection with a stock split, stock dividend, recapitalization, or similar event or adjustment. In addition, an indeterminate number of shares of common stock are registered hereunder that may be issued upon conversion of or exchange for any convertible preferred stock or debt securities, or upon exercise of any warrant. | |||||||||
(4) | In no event will the aggregate initial offering price of all securities issued from time to time by the registrant pursuant to this registration statement exceed $75,000,000 or the equivalent thereof in one or more foreign currencies, foreign currency units or composite currencies, excluding accrued interest, if any, on any debt securities issued under the registration statement. | |||||||||
The registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment that specifically states that this registration statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act or until the registration statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
SUBJECT TO COMPLETION, DATED FEBRUARY 10, 2020
PROSPECTUS
$75,000,000

Common Stock
Preferred Stock
Debt Securities
Warrants
Units
Debt Securities
Warrants
Units
We may from time to time offer, in one or more series or classes, separately or together, and in amounts, at prices and on terms to be set forth in one or more supplements to this prospectus, the following securities:
• | shares of our common stock, par value $0.01 per share; |
• | shares of our preferred stock (which we may issue in one or more classes or series), par value $0.01 per share; |
• | debt securities; |
• | warrants to purchase shares of common stock or preferred stock or debt securities; or |
• | units consisting of two or more of the foregoing. |
We refer to the common stock, preferred stock, debt securities, warrants and units, collectively, as the “securities” in this prospectus. We may offer, issue and sell the securities at an aggregate public offering price that will not exceed $75,000,000.
We will provide the specific amount, price and terms of any securities we may offer in one or more supplements to this prospectus. You should carefully read this prospectus and the applicable prospectus supplement, as well as the documents incorporated or deemed to be incorporated by reference in this prospectus, before you purchase any of the securities offered hereby. This prospectus may not be used to offer and sell any securities unless accompanied by a prospectus supplement describing the amount of securities being offered and the terms of the offering of those securities.
We may offer and sell these securities to or through one or more underwriters, dealers, and agents, or directly to purchasers on a continuous or delayed basis. We reserve the sole right to accept, and together with any underwriters, dealers and agents, reserve the right to reject, in whole or in part, any proposed purchase of securities. The names of any underwriters, dealers or agents involved in the sale of any securities, the specific manner in which they may be offered and any applicable commissions or discounts will be set forth in the prospectus supplement covering the sale of those securities.
Our common stock is listed on The Nasdaq Capital Market under the symbol “BBI.” On February 7, 2020, the last reported sale price of our common stock on The Nasdaq Capital Market was $1.53.
As of February 7, 2020, the aggregate market value of our outstanding common stock held by non-affiliates was approximately $10,472,074, which was calculated based on 6,844,493 shares of outstanding common stock held by non-affiliates and a price per share of $1.53. Pursuant to General Instruction I.B.6 of Form S-3, in no event will we sell the shelf securities in a public primary offering with a value exceeding more than one-third of the aggregate market value of our voting and non-voting common stock held by non-affiliates in any 12-month period as long as the aggregate market value of our outstanding voting and non-voting common stock held by non-affiliates is less than $75 million. We have sold no securities pursuant to General Instruction I.B.6 of Form S-3 during the 12 calendar months prior to, and including, the date of this prospectus.
INVESTING IN OUR SECURITIES INVOLVES RISKS. SEE THE “RISK FACTORS” BEGINNING ON PAGE 6 OF THIS PROSPECTUS AND ANY SIMILAR SECTION CONTAINED IN THE APPLICABLE PROSPECTUS SUPPLEMENT OR ANY DOCUMENTS THAT ARE INCORPORATED BY REFERENCE INTO THIS PROSPECTUS BEFORE INVESTING IN OUR SECURITIES.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the adequacy or accuracy of this prospectus. Any representation to the contrary is a criminal offense.
The date of this prospectus is February 10, 2020.
TABLE OF CONTENTS
ABOUT THIS PROSPECTUS | |
CAUTIONARY NOTE CONCERNING FORWARD-LOOKING STATEMENTS | |
SUMMARY INFORMATION | |
RISK FACTORS | |
BUSINESS | |
USE OF PROCEEDS | |
DESCRIPTION OF SECURITIES WE MAY OFFER | |
DESCRIPTION OF CAPITAL STOCK | |
DESCRIPTION OF DEBT SECURITIES | |
DESCRIPTION OF WARRANTS | |
DESCRIPTION OF UNITS | |
PLAN OF DISTRIBUTION | |
INFORMATION INCORPORATED BY REFERENCE | |
WHERE YOU CAN FIND MORE INFORMATION | |
LEGAL MATTERS | |
EXPERTS | |
i
ABOUT THIS PROSPECTUS
This prospectus is part of a registration statement on Form S-3 that we filed with the Securities and Exchange Commission (the “SEC”), using a “shelf” registration process for the delayed offering and sale of securities pursuant to Rule 415 under the Securities Act of 1933, as amended (the “Securities Act”). Under this shelf registration process, we may, over time, offer and sell any combination of the securities described in this prospectus in one or more offerings, up to a total dollar amount of $75,000,000. This prospectus provides you with a general description of the securities that may be offered. Each time we offer securities under this prospectus, we will provide a prospectus supplement or other offering materials that will contain specific information about the terms of that offering. We are subject to the provisions of General Instruction I.B.6 of the General Instructions to Form S-3, which provide that as long as the aggregate market value of our outstanding voting and non-voting common stock held by non-affiliates is less than $75 million, then the aggregate market value of securities sold by us or on our behalf on Form S-3 during the 12-month period immediately prior to, and including, the sale, is no more than one-third of the aggregate market value of our voting and non-voting common stock held by non-affiliates.
We may also add, update or change information contained in this prospectus by means of a prospectus supplement or by incorporating by reference information that we file or furnish to the SEC. The registration statement that we filed with the SEC includes exhibits that provide more detail on the matters discussed in this prospectus. If the information in this prospectus is inconsistent with a prospectus supplement, you should rely on the information in that prospectus supplement. Please carefully read this prospectus and any prospectus supplement, together with the additional information described under the headings “Where You Can Find More Information” and “Information Incorporated by Reference” before purchasing any securities.
You should rely only on the information contained or incorporated by reference in this prospectus, any prospectus supplement and any issuer free writing prospectus. “Incorporated by reference” means that we can disclose important information to you by referring you to another document filed separately with the SEC. We have not authorized any other person to provide you with different information. If anyone provides you with different information, you should not rely on it. We are not making an offer of these securities in any state or jurisdiction where the offer is not permitted. You should only assume that the information in this prospectus or in any prospectus supplement or issuer free writing prospectus is accurate only as of their respective dates. Our business, financial condition, results of operations and prospects may have changed since those dates.
In this prospectus, “Brickell,” the “Company,” “we,” “us” and “our” refer to Brickell Biotech, Inc. and its consolidated subsidiaries, except where the context otherwise requires.
1
CAUTIONARY NOTE CONCERNING FORWARD‑LOOKING STATEMENTS
This prospectus and any accompanying prospectus supplement, including the documents incorporated by reference into this prospectus and any accompanying prospectus supplement, contain forward‑looking statements within the meaning of Section 27A of the Securities Act and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). These forward-looking statements are intended to provide management’s current expectations or plans for future operating and financial performance based on assumptions currently believed to be valid. Forward-looking statements can be identified by the use of words such as “believe,” “expect,” “assume,” “expectations,” “plans,” “strategy,” “prospects,” “estimate,” “project,” “target,” “anticipate,” “will,” “may,” “should,” “see,” “guidance,” “confident” and other words of similar meaning in connection with a discussion of future operating or financial performance. All forward-looking statements involve risks, uncertainties and other factors that may cause actual results to differ materially from those expressed or implied in the forward-looking statements. Risks, uncertainties and other factors that could cause actual results to differ from these forward-looking statements include, but are not limited to, risks and uncertainties detailed in the section titled “Risk Factors” beginning on page 6. The statements made in this prospectus and any accompanying prospectus supplement, including the documents incorporated by reference into this prospectus and any accompanying prospectus supplement, regarding the following subject matters are forward-looking by their nature:
• | impact of our current litigation with Bodor Laboratories, Inc. (“Bodor”) relating to our right to continue to develop sofpironium bromide; |
• | our expected cash position and our ability to obtain financing in the future on satisfactory terms or at all; |
• | estimates of our expenses and capital requirements; |
• | expectations regarding the successful development, regulatory approval and commercialization of sofpironium bromide and our early stage product candidates; |
• | expectations regarding the results and timing of results of clinical trials for sofpironium bromide and our other product candidates; |
• | expectations regarding the potential market size, opportunity and growth potential for sofpironium bromide and our early stage product candidates; |
• | expectations regarding the degree of physician and patient adoption, reimbursement and use of sofpironium bromide following approval, if received; |
• | our relationship with, and expectations of, our product development partners and licensors; |
• | expectations regarding the safety, efficacy and quality of our early stage product candidates; |
• | the timing or likelihood of regulatory filings and approvals; |
• | the implementation of our business model, strategic plans for our business, product candidates and technology; |
• | the scope of protection we are able to acquire, establish, maintain and enforce for intellectual property rights covering our product candidates and technology; |
• | developments relating to our competitors; and |
• | any future litigation or threat of litigation. |
The preceding list is not intended to be an exhaustive list of all forward-looking statements in this prospectus and any accompanying prospectus supplement. You should read this prospectus and any accompanying prospectus supplement with the understanding that actual future results, levels of activity, performance and achievements may be materially different
2
from what is currently expected. We qualify all of the forward-looking statements by these cautionary statements. Additional factors that could cause results to differ materially from those described above can be found in the reports and information that we file with the SEC from time to time.
3
SUMMARY INFORMATION
This summary does not contain all the information that you should consider before investing in our Company. You should carefully read the entire prospectus and any accompanying prospectus supplement, including all documents incorporated by reference herein and therein.
Company Overview
We are a clinical-stage pharmaceutical company focused on the development of innovative and differentiated prescription therapeutics for the treatment of debilitating skin diseases. Our pipeline consists of potential novel therapeutics for hyperhidrosis, cutaneous T-cell lymphoma, psoriasis, and other prevalent dermatological conditions. Our executive management team and board of directors bring extensive experience in product development and global commercialization, having served in leadership roles at large global pharmaceutical companies and biotechs that have developed and/or launched successful products, including several that were first-in-class and/or achieved iconic status, such as Cialis®, Taltz®, Gemzar®, Prozac®, Cymbalta® and Juvederm®.
Our pivotal Phase 3-ready clinical-stage product candidate, sofpironium bromide, is a proprietary new molecular entity. It belongs to a class of medications called anticholinergics. Anticholinergics block the action of acetylcholine, a chemical that transmits signals within the nervous system that are responsible for a range of bodily functions, including activation of the sweat glands. Sofpironium bromide was retrometabolically designed. Retrometabolic drugs are designed to exert their action topically and are potentially rapidly metabolized once absorbed into the blood. This proposed mechanism of action may allow for highly effective doses to be used while limiting systemic side effects. We are developing sofpironium bromide as a potential best-in-class, self-administered, once-daily, topical therapy for the treatment of primary axillary hyperhidrosis. Hyperhidrosis is a life-altering condition of sweating beyond what is physiologically required to maintain normal thermal regulation. It is believed to be caused by an overactive cholinergic response of the sweat glands, and affects an estimated 15.3 million, or 4.8%, of the U.S. population. According to a 2016 update on the prevalence and severity of hyperhidrosis in the United States by Doolittle et al., axillary (underarm) hyperhidrosis, which is the targeted first potential indication for sofpironium bromide, is the most common occurrence of hyperhidrosis, affecting approximately 65% of patients in the United States or an estimated 10 million individuals.
We and our development partner in Asia, Kaken Pharmaceutical Co. Ltd. (“Kaken”), have conducted 19 clinical trials of sofpironium bromide gel that encompass over 1,200 subjects in the United States and Japan. These trials evaluated the potential safety, tolerability, pharmacokinetics (“PK”), and efficacy of sofpironium bromide gel in adult and pediatric primary axillary hyperhidrosis patients and healthy adult subjects. Under our License, Development and Commercialization Agreement with Kaken, dated March 31, 2015 (the “Kaken Agreement”), in exchange for paying us an upfront, nonrefundable payment, we granted Kaken the exclusive right to develop, manufacture and commercialize sofpironium bromide in Japan and certain other Asian countries. In March 2019, Kaken completed a Phase 3 trial in patients with primary axillary hyperhidrosis in Japan, achieving statistical significance on all primary and secondary endpoints. In January 2020, we announced that Kaken submitted a new drug application for approval of manufacturing and marketing for sofpironium bromide in Japan for primary axillary hyperhidrosis.
Based on the positive results of the clinical trials for sofpironium bromide to date, we intend to initiate two pivotal Phase 3 clinical trials in up to 350 subjects per trial with primary axillary hyperhidrosis in the United States, subject to obtaining substantial additional funding and the successful resolution of our dispute with Bodor and Dr. Nicholas S. Bodor (collectively, the “Bodor Plaintiffs”) as described below. Assuming the results of the Phase 3 clinical trials are favorable, we plan thereafter to submit a New Drug Application (“NDA”) to the U.S. Food and Drug Administration (“FDA”) for the treatment of hyperhidrosis by sofpironium bromide.
On October 23, 2019, Bodor notified us of its purported termination of the license agreement we entered into with Bodor, dated December 15, 2012, as amended by Amendment No. 1 to the License Agreement, effective as of October 21, 2013, and Amendment No. 2 to the License Agreement, effective as of March 31, 2015 (the “License Agreement”). In connection with this attempt, on October 23, 2019, the Bodor Plaintiffs filed a complaint (the “Bodor Complaint”) against us in the United States District Court for the Southern District of Florida. The Bodor Complaint alleges damages incurred by the Bodor Plaintiffs in connection with our alleged material breach of the License Agreement and requests a declaratory judgment that the attempted termination of the License Agreement by Bodor be permitted. As required by the License Agreement, we initiated an arbitration proceeding and then subsequently a mediation with the Bodor Plaintiffs to resolve the dispute. In our arbitration demand, we countersued the Bodor Plaintiffs for damages caused by them to us, both under
4
contract and due to their tortious interference with our business relations. We asked the district court to dismiss or stay the Bodor Complaint. The judge administratively closed the case pending arbitration. We believe that the claims and assertions made by the Bodor Plaintiffs are in substance without merit. Following an unsuccessful mediation, we and the Bodor Plaintiffs commenced arbitration. Pending successful resolution of this dispute and obtaining substantial additional funding, we intend to conserve our resources. The advancement of the Phase 3 clinical trials for sofpironium bromide have been negatively impacted by these developments and will require substantial additional funds. We have taken, and expect to continue to take, actions to reduce our cash spend, including delaying the start of the clinical trials and/or staff reductions.
Our second product candidate, BBI-3000, is a selective, potentially highly tolerable and potent novel retinoid X receptor (“RXR”) agonist that we are evaluating for the development in cutaneous T-cell lymphoma (“CTCL”) as a potential oral treatment. Retinoids are derivatives of vitamin A that play a pivotal role in a diverse group of biologic processes including, but not limited to, cellular proliferation, differentiation, apoptosis, and development. The biological activity and tolerability of retinoids depends in part on the binding availability to retinoic acid and RXR receptors. There are several topical and oral retinoids currently on the market that have shown efficacy in the treatment of several skin conditions, such as CTCL (e.g., bexarotene/Targretin®), acne and psoriasis (e.g., tazarotene, adapalene and tretinoin). BBI-3000 has been well tolerated in two Phase 1 studies (a single dose study and a multiple dose study) conducted by the National Cancer Institute (“NCI”) in healthy volunteers. There is an ongoing Phase 1b trial being conducted by the NCI to assess the biological effect of BBI-3000 on early stage breast cancer.
Our third product candidate, BBI-6000, is a novel retinoic acid-related orphan nuclear receptor gamma (“RORg”) inhibitor that we are developing for the topical treatment of mild-to-moderate psoriasis. RORg inhibition targets the pathway of a validated cytokine (“IL-17”) that has been implicated in the pathogenesis of psoriasis. Monoclonal antibodies targeting IL‑17 have recently shown significant efficacy in the treatment of psoriasis, and we are planning to develop BBI‑6000 as a topically applied, potent and selective small-molecule therapeutic targeting this pathway. BBI-6000 is currently in the preclinical stages of development.
Merger of Brickell Biotech, Inc. and Vical Incorporated
On August 31, 2019, the Delaware corporation formerly known as “Vical Incorporated”, completed a reverse merger transaction in accordance with the terms and conditions of the Agreement and Plan of Merger and Reorganization, dated as of June 2, 2019, as amended by Amendment No. 1 to Agreement and Plan of Merger and Reorganization, dated August 20, 2019, and as further amended on August 30, 2019 (the “Merger Agreement”), by and among Vical Incorporated (“Vical”), Brickell and Victory Subsidiary, Inc., a wholly-owned subsidiary of Vical formed in connection with the merger (the “Merger Sub”), pursuant to which the Merger Sub merged with and into Brickell, with Brickell surviving the merger as a wholly-owned subsidiary of Vical (the “Merger”). Additionally, on August 31, 2019, immediately after the completion of the Merger, the Company changed its name from “Vical Incorporated” to “Brickell Biotech, Inc.” On August 31, 2019, in connection with, and prior to the consummation of the Merger, Vical effected a reverse stock split of its common stock, par value $0.01 per share, at a ratio of 1-for-7.
Concurrent with the execution of the Merger Agreement, we entered into a Funding Agreement, dated as of June 2, 2019 (the “Funding Agreement”), with NovaQuest Co-Investment Fund X, L.P. (“NovaQuest”) pursuant to which NovaQuest committed partially to fund our expenses relating to certain product development activities. As a result of the dispute with the Bodor Plaintiffs, on November 25, 2019, Brickell Subsidiary, Inc., our wholly-owned subsidiary, and NovaQuest entered into a Settlement and Termination Agreement (the “Settlement and Termination Agreement”) effectively terminating the Funding Agreement. NovaQuest agreed to cancel and surrender the warrant it previously received in connection with the Funding Agreement, and we repaid NovaQuest the $5.6 million advance previously made by NovaQuest in addition to accrued interest. Subject to the mutual indemnity included in the Settlement and Termination Agreement, NovaQuest agreed to waive any and all of our further obligations (including any and all future milestone payments and royalties owed to NovaQuest) and each party agreed to release any and all claims against the other party in respect of the Funding Agreement.
Our common stock trades on The Nasdaq Capital Market under the symbol “BBI.” Our principal executive offices are located at 5777 Central Avenue, Suite 102, Boulder, Colorado 80301, our telephone number is (720) 505-4755 and our corporate website address is www.brickellbio.com. Our website and the information contained on or accessible through our website are not part of this document. We have included our website address in this prospectus solely as an inactive textual reference.
5
RISK FACTORS
An investment in our securities involves risks. We urge you to carefully consider all of the information contained in or incorporated by reference in this prospectus and other information which may be incorporated by reference in this prospectus or any prospectus supplement as provided under “Information Incorporated by Reference.” This prospectus also contains forward-looking statements that involve risks and uncertainties. Please read “Cautionary Note Concerning Forward-Looking Statements.” Our actual results could differ materially from those anticipated in the forward-looking statements as a result of certain factors, including the risks described below or in any prospectus supplement and in the documents incorporated by reference into this prospectus or any prospectus supplement. If any of these risks occur, this could expose us to liability, and our business, financial condition or results of operation could be adversely affected. As a result, you could lose all or part of your investment.
Risks Related to the Development, Commercialization and Regulatory Approval of Our Investigational Drug, Sofpironium Bromide
We are currently involved in litigation with the Bodor Plaintiffs relating to our right to continue to develop sofpironium bromide.
On October 23, 2019, Bodor notified us of its purported termination of the License Agreement. Bodor alleges that we materially breached the License Agreement resulting in its termination. In connection with this purported termination, on October 23, 2019, the Bodor Plaintiffs filed the Bodor Complaint against us in the United States District Court for the Southern District of Florida. The Bodor Complaint alleges damages incurred by the Bodor Plaintiffs in connection with our alleged material breach of the License Agreement. The Bodor Complaint seeks: (i) a declaratory judgment that the termination of the License Agreement by the Bodor Plaintiffs was valid and enforceable; (ii) an injunction requiring us to cease and desist use of the Bodor Plaintiffs’ intellectual property; and (iii) damages for breach of contract and breach of the covenant of good faith and fair dealing.
On October 30, 2019, we initiated an arbitration proceeding pursuant to Article 9 of the License Agreement with the American Arbitration Association (“AAA”), in Florida against the Bodor Plaintiffs. This arbitration seeks a declaratory judgment that the purported termination of the License Agreement by the Bodor Plaintiffs was invalid and unenforceable and asserts (i) a claim for breach of the License Agreement against the Bodor Plaintiffs, and (ii) a claim against the Bodor Plaintiffs for tortious interference with our business relations. On October 30, 2019, we concurrently filed with the United States District Court for the Southern District of Florida a motion to dismiss (or stay) the complaint brought against us by the Bodor Plaintiffs described above.
In its response to our motion to dismiss or, alternatively, stay the litigation, Bodor agreed to comply with the License Agreement terms, which require mediation and, if necessary, arbitration to resolve the dispute. Bodor also asked the court to stay the federal judicial proceedings against us pending mediation, and, if necessary, final, conclusive and binding arbitration. We proceeded with mediation on December 10, 2019, which was unsuccessful. As a result, we recommenced the arbitration proceeding with the Bodor Plaintiffs to conclude the dispute. In December 2019, we recorded an estimated loss contingency of $1.0 million for this matter and will continue to evaluate the adequacy of this estimate as the matter develops.
As a result of these matters, the timeline and funding for our Phase 3 clinical trials in subjects with primary axillary hyperhidrosis in the United States has been negatively impacted. In addition, we have been required to devote substantial financial resources to address the Bodor Complaint. Arbitration and litigation are expensive and may likely be time-consuming and divert management’s attention and our resources away from other clinical development activities.
The outcome of arbitration or litigation is inherently uncertain. If one or more of the legal claims made by the Bodor Plaintiffs were resolved against us, we may become subject to additional litigation claims, and/or our ability to continue to develop sofpironium bromide and our financial condition and operating results could be materially adversely affected. While we maintain insurance coverage for certain types of claims, such insurance coverage may be insufficient to cover all losses or all types of claims that may arise.
Unless we are able to negotiate an acceptable settlement with the Bodor Plaintiffs in the near future, we may have difficulty retaining our key employees who have extensive knowledge and experience in developing and commercializing pharmaceutical products. We may be required to undertake a reduction in personnel and to minimize our expenses until such time as we prevail in our arbitration with the Bodor Plaintiffs. To the extent that our cash resources are depleted as a result of
6
the mediation and arbitration-related expenses, we may be required to consider other measures. Even if we reach a favorable settlement with the Bodor Plaintiffs, there is no assurance that we will be able to raise the capital necessary to advance sofpironium bromide through the Phase 3 clinical trials on the timetable we contemplated. To the extent that we are unable to raise capital sufficient to fund our clinical development, there may be substantial doubt regarding our ability to remain a going concern.
Our business depends on the successful financing, clinical development, regulatory approval and commercialization of sofpironium bromide.
The success of our business, including our prospective ability to finance our operations and generate revenue, primarily depends on the successful development, regulatory approval and commercialization of sofpironium bromide, at least in the United States. The clinical and commercial success of sofpironium bromide depends on a number of factors, including but not limited to the following:
• | timely and successful completion of Phase 3 clinical trials in the United States not yet initiated, which may be significantly delayed particularly in light of the Bodor Complaint, or costlier than we currently anticipate and/or produce results that do not achieve the endpoints of the trials or which are ultimately deemed not to be clinically meaningful; |
• | whether we are required by the FDA or similar foreign regulatory agencies to conduct additional clinical trials beyond those currently planned to support the approval and commercialization of sofpironium bromide; |
• | achieving and maintaining, and, where applicable, ensuring that our third-party contractors achieve and maintain, compliance with our and their contractual obligations and with all regulatory and legal requirements applicable to sofpironium bromide; |
• | ability of third parties with which we contract to manufacture consistently adequate clinical trial and commercial supplies of sofpironium bromide, to remain in good standing with regulatory agencies and to develop, validate and maintain or supervise commercially viable manufacturing processes that are compliant with FDA-regulated Current Good Manufacturing Practices (“cGMPs”) and the product’s package insert; |
• | a continued acceptable safety profile during clinical development and following approval of sofpironium bromide; |
• | ability to obtain favorable labeling for sofpironium bromide through regulators that allows for successful commercialization, given the drug may be marketed only to the extent approved by these regulatory authorities (unlike with most other industries); |
• | ability to commercialize sofpironium bromide successfully in the United States and internationally, if approved for marketing, sale and distribution in such countries and territories, whether alone or in collaboration with Kaken or others; |
• | acceptance by physicians, insurers and payors, and patients of the quality, benefits, safety and efficacy of sofpironium bromide, if approved, including relative to alternative and competing treatments and the next best standard of care; |
• | existence of a regulatory and legal environment conducive to the success of sofpironium bromide; |
• | ability to price sofpironium bromide to recover our development costs and generate a satisfactory profit margin; and |
7
• | our ability and our partners’ ability to establish and enforce intellectual property rights in and to sofpironium bromide, including but not limited to patents and licenses. |
If we do not achieve one or more of these factors, many of which are beyond our reasonable control, in a timely manner or at all, and with adequate financing, we could experience significant delays or an inability to obtain regulatory approvals or commercialize sofpironium bromide. Even if regulatory approvals are obtained, we may never be able to successfully commercialize sofpironium bromide. Accordingly, we cannot assure you that we will be able to generate sufficient revenue through the sale of sofpironium bromide, or any current primary asset, to continue our business.
We have never conducted a Phase 3 clinical trial ourselves and may be unable to successfully do so for sofpironium bromide.
The conduct of a Phase 3 clinical trial is a long, expensive, complicated, uncertain and highly regulated process. Although our employees have conducted successful Phase 2 and Phase 3 clinical trials in the past across many therapeutic areas while employed at other companies, we as a company have not conducted a Phase 3 pivotal clinical trial, and as a result, we may require more time and incur greater costs than we anticipate. We commenced a Phase 3 long-term safety study for sofpironium bromide gel in the third quarter of 2018 and intend to initiate two pivotal Phase 3 clinical trials in subjects with primary axillary hyperhidrosis in the United States, subject to the successful resolution of our dispute with the Bodor Plaintiffs and obtaining substantial additional funding. Failure to commence or complete, or delays in, our planned clinical trials would prevent us from, or delay us in, obtaining regulatory approval of and commercializing sofpironium bromide and could prevent us from, or delay us in, receiving development- or regulatory-based milestone payments and commercializing sofpironium bromide gel for the treatment of hyperhidrosis, which would adversely impact our financial performance, as well as put us in potential breach of material contracts for the licensing and development of sofpironium bromide, subjecting us to significant contract liabilities, including but not limited to loss of rights in and to sofpironium bromide.
Clinical drug development for sofpironium bromide is very expensive, time-consuming and uncertain.
Clinical development for sofpironium bromide is very expensive, time-consuming, difficult to design and implement, and its outcome is inherently uncertain. Most product candidates that commence clinical trials are never approved by regulatory authorities for commercialization and of those that are approved many do not cover their costs of development or ever generate a profit. In addition, we, any partner with which we currently or may in the future collaborate, the FDA, a local or central institutional review board (“IRB”), or other regulatory authorities, including state and local agencies and counterpart agencies in foreign countries, may suspend, delay, extend, require modifications or add additional requirements to or terminate our clinical trials at any time.
In the case of sofpironium bromide, we are seeking to deliver sufficient concentrations of the active pharmaceutical ingredient (“API”) absorbed from the skin surface through the skin barrier to the targeted dermal tissue to achieve the intended therapeutic effect, in this case treatment of hyperhidrosis. The topical route of administration may involve new dosage forms, which can be difficult to develop and manufacture and may raise novel regulatory issues and result in development or review delays or inability to get the investigational drug approved for use.
Use of patient-reported outcome assessments (“PROs”) and gravimetric assessments in sofpironium bromide clinical trials may delay or adversely impact the development of sofpironium bromide gel or clinical trial results or increase our development costs.
Due to the difficulty of objectively measuring the symptoms of hyperhidrosis in a clinical trial, which is the primary target of treatment for sofpironium bromide, PROs will have an important role in the development and regulatory approval of sofpironium bromide. PROs involve patients’ own subjective assessments of efficacy, and this subjectivity increases the uncertainty of determining and achieving clinical endpoints and obtaining regulatory approval. Such assessments can be influenced by factors outside of our reasonable control and can vary widely from day to day for a particular patient, and from patient to patient and site to site within a clinical trial, notwithstanding that regulators may or may not accept PROs as part of the drug approval process. Additionally, gravimetric assessments of sweat production, another key clinical endpoint, may vary significantly for a particular patient, and from patient to patient and site to site within a clinical trial or between separate clinical trials. The reduction, if any, in a patient’s gravimetric sweat production has the potential for significant variability and uncertain outcomes. This potential for variability and uncertain outcomes may adversely impact our ability to achieve statistical significance on our primary and secondary endpoints or may provide us with initial or subsequent results that are ultimately deemed not to be clinically meaningful or that do not result in regulatory approval.
8
Sofpironium bromide may cause undesirable side effects or have other unexpected properties that could delay or prevent its regulatory approval, limit the commercial profile of an approved label, or result in post-approval regulatory action.
Unforeseen side effects from sofpironium bromide could arise either during clinical development or, if approved, after it has been marketed. Undesirable side effects caused by sofpironium bromide could cause us, any partners with which we may collaborate, or regulatory authorities to interrupt, extend, modify, delay or halt clinical trials and could result in a more restrictive or narrower product label or the delay or denial of regulatory approval by the FDA or comparable foreign authorities.
Results of clinical trials could reveal a high and unacceptable severity and prevalence of side effects. In such an event, trials could be suspended or terminated, and the FDA or comparable foreign regulatory authorities could order us to cease further development of or deny approval of sofpironium bromide for any or all targeted indications. The drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in product liability claims. Any of these occurrences may expose us to liability or harm our business, financial condition, operating results and prospects.
Additionally, if we or others identify undesirable side effects, or other previously unknown problems, caused by sofpironium bromide after obtaining U.S. or foreign regulatory approval, a number of potentially negative consequences could result, which could prevent us or our potential partners from achieving or maintaining market acceptance of sofpironium bromide and could substantially increase the costs of commercializing sofpironium bromide, potentially even leading to recall of the drug.
Kaken substantially controls the development of sofpironium bromide in Japan and certain other Asian countries and may make decisions regarding product development, regulatory strategy and commercialization that may not be in our best interests. Kaken may be unable to obtain positive approval of the drug in Asian markets.
The Kaken Agreement granted Kaken an exclusive Japan license and certain rights to additional Asian countries to develop and commercialize sofpironium bromide. Under the terms of the agreement, as amended, we received an up-front payment, development milestones and research and development payments and are eligible to receive future milestones and a royalty on net sales.
Kaken has final decision-making authority for the overall regulatory, development and commercialization strategy for sofpironium bromide, market access activities, pricing and reimbursement activities, promotion, distribution, packaging, sales and safety and pharmacovigilance in Japan and certain other Asian countries. In exercising its final decision-making authority in such territories, Kaken may make decisions regarding product development or regulatory strategy based on its determination of how best to preserve and extend regulatory approvals in these territories for sofpironium bromide, which may delay or prevent achieving regulatory approval for sofpironium bromide in Kaken’s territories, as well as by us in the United States and the other territories where we maintain exclusive rights. Additionally, Kaken is responsible for conducting certain nonclinical and API (chemistry, manufacturing and controls) -related activities that will be required for FDA approval in the United States, and as a result, we are reliant on Kaken to execute successfully, in a timely and efficient manner, such activities on our behalf. To the extent Kaken experiences delays and/or difficulties in performing its development activities, this could prevent or cause substantial delays in our ability to seek approval for sofpironium bromide gel in the United States and other territories in which we maintain exclusive rights. We will not receive additional milestone or other payments from Kaken if Kaken is not successful in its development activities.
If we or any partners with which we may collaborate to market and sell sofpironium bromide are unable to achieve and maintain insurance coverage and adequate levels of reimbursement for this compound following regulatory approval and usage by patients, our commercial success may be hindered severely.
If sofpironium bromide only becomes available by prescription, successful sales by us or by any partners with which we collaborate may depend on the availability of insurance coverage and adequate reimbursement from third-party payors as patients would then be forced to pay for the drug out-of-pocket if coverage and associated reimbursement is denied. Patients who are prescribed medicine for the treatment of their conditions generally rely on third-party payors to reimburse all or part of the costs associated with their prescription drugs. The availability of coverage and adequate reimbursement from governmental healthcare programs, such as Medicare and Medicaid in the United States, and private third-party payors is often critical to new product acceptance regardless of how well the product works. Coverage decisions may depend on clinical and economic standards that disfavor new drug products when more established or lower-cost therapeutic alternatives
9
are already available or subsequently become available, even if these alternatives are not as safe and effective, or may be affected by the budgets and demands on the various entities responsible for providing health insurance to patients who will use sofpironium bromide. If insurers and payors decide that hyperhidrosis itself is not a disease they are willing to extend coverage to, which could happen if they only think the treatment improves quality of life, then coverage and reimbursement for sofpironium bromide may be denied, or at least severely restricted. In this case, patients would be forced to pay for sofpironium bromide out-of-pocket for cash, which they may not be willing or able to do. Even if we obtain coverage for sofpironium bromide, the resulting reimbursement payment rates might not be adequate or may require co-payments that patients find unacceptably high. Patients may not use sofpironium bromide unless coverage is provided and reimbursement is adequate to cover a significant portion of the cost of sofpironium bromide.
In addition, the market for sofpironium bromide will depend significantly on access to third-party payors’ drug formularies or lists of medications for which third-party payors provide coverage and reimbursement. The industry competition to be included in such formularies often leads to downward pricing pressures on pharmaceutical companies and there may be time limitations on when a new drug may even be eligible for formulary inclusion. Also, third-party payors may refuse to include sofpironium bromide in their formularies or otherwise restrict patient access to sofpironium bromide when a less costly generic equivalent or other treatment alternative is available in the discretion of the formulary.
Third-party payors, whether foreign or domestic, or governmental or commercial, are developing increasingly sophisticated methods of controlling healthcare costs. In addition, in the United States, although private third-party payors tend to follow Medicare and Medicaid practices, no uniform or consistent policy of coverage and reimbursement for drug products exists among third-party payors. Therefore, coverage and reimbursement for drug products can differ significantly from payor to payor as well as state to state. Consequently, the coverage determination process is often uncertain and a time-consuming and costly process that must be played out across many jurisdictions and different entities and which will require us to provide scientific, clinical and health economics support for the use of sofpironium bromide compared to current alternatives and do so to each payor separately, with no assurance that coverage and adequate reimbursement will be obtained and in what amount or time frame.
Further, we believe that future coverage and reimbursement likely will be subject to increased restrictions both in the United States and in international markets. Third-party coverage and reimbursement for sofpironium bromide may not be available or adequate in either the United States or international markets, which could harm our business, financial condition, operating results and prospects.
Even if sofpironium bromide obtains regulatory approval, it may fail to achieve the broad degree of physician and patient adoption and use necessary for commercial success.
The commercial success of sofpironium bromide, if approved, will depend significantly on the broad adoption and use of it by physicians and patients for approved indications, and may not be commercially successful even though the drug is shown to be safe and effective. The degree and rate of physician and patient adoption of sofpironium bromide, if approved, especially in the United States, will depend on a number of factors, including but not limited to:
• | patient demand for approved products that treat hyperhidrosis; |
• | our ability to market and sell the drug, including through direct-to-consumer advertising and non-traditional sales strategies; |
• | the safety and effectiveness of sofpironium bromide, and ease of use, compared to other available hyperhidrosis therapies, whether approved or used by physicians off-label; |
• | the availability of coverage and adequate reimbursement from managed care plans and other healthcare payors for sofpironium bromide; |
• | the cost of treatment with sofpironium bromide in relation to alternative hyperhidrosis treatments and willingness to pay for sofpironium bromide, if approved, on the part of patients; |
10
• | overcoming physician or patient biases toward particular therapies for the treatment of hyperhidrosis and achieving acceptance by physicians, major operators of clinics and patients of sofpironium bromide as a safe, effective and economical hyperhidrosis treatment; |
• | patients’ perception of hyperhidrosis as a disease and one for which medical treatment may be appropriate and a prescription therapy may be available; |
• | insurers’ and physicians’ willingness to see hyperhidrosis as a disease worth treating and for which reimbursement will be made available for treatment; |
• | proper administration of sofpironium bromide; |
• | patient satisfaction with the results and administration of sofpironium bromide and overall treatment experience; |
• | limitations or contraindications, warnings, precautions or approved indications for use different than those sought by us that are contained in any final FDA-approved labeling for sofpironium bromide; |
• | any FDA requirement to undertake a risk evaluation and mitigation strategy; |
• | the effectiveness of our sales, marketing, pricing, reimbursement and access, government affairs, legal, medical and distribution efforts; |
• | adverse publicity about sofpironium bromide or favorable publicity about competitive products; |
• | new government regulations and programs, including price controls and/or public or private institutional limits or prohibitions on ways to commercialize drugs, such as increased scrutiny on direct-to-consumer advertising of pharmaceuticals or restrictions on sales representatives to market pharmaceuticals; and |
• | potential product liability claims or other product-related litigation or litigation related to licensing and or other commercial matters associated with sofpironium bromide. |
If sofpironium bromide is approved for use but fails to achieve the broad degree of physician and patient adoption necessary for commercial success, our operating results and financial condition will be adversely affected, which may delay, prevent or limit our ability to generate revenue and continue our business.
Sofpironium bromide, if approved, will face significant competition and its failure to compete effectively may prevent it from achieving significant market penetration.
The pharmaceutical industry is characterized by rapidly advancing technologies, intense competition, less effective patent terms, and a strong emphasis on developing newer, fast-to-market proprietary therapeutics. Numerous companies are engaged in the development, patenting, manufacturing and marketing of healthcare products competitive with those that we are developing, including sofpironium bromide. We face competition from a number of sources, such as pharmaceutical companies, generic drug companies, biotechnology companies and academic and research institutions, many of which have greater financial resources, marketing capabilities, sales forces, manufacturing capabilities, research and development capabilities, regulatory expertise, clinical trial expertise, intellectual property portfolios, more international reach, experience in obtaining patents and regulatory approvals for product candidates and other resources than us. Some of the companies that offer competing products also have a broad range of other product offerings, large direct sales forces and long-term customer relationships with our target physicians, which could inhibit our market penetration efforts. In addition, sofpironium bromide, if approved, may compete with other dermatological products, including over-the-counter treatments, for a share of some patients’, or payors’, discretionary budgets and for physicians’ attention within their clinical practices.
We anticipate that sofpironium bromide would compete with other therapies currently used for hyperhidrosis, including but not limited to:
11
• | Self-Administered Treatments. Self-administered treatments, such as OTC and prescription topical antiperspirants, and Qbrexza® (glycopyrronium) 2.4% topical cloths. Oral and compounded topical anticholinergics also may be used off-label. |
• | Non-Surgical Office-Based Procedures. Office-based procedures have been approved by the FDA for certain uses and which may be used, on-or off-label, to treat hyperhidrosis, including intradermal injections of BOTOX®, marketed by Allergan plc., and MiraDry®, a microwave-based treatment marketed by Miramar Labs, Inc. |
• | Surgical Treatments. Surgical treatments include techniques for the removal of sweat glands, such as excision, curettage and liposuction. Surgical procedures, such as endoscopic thoracic sympathectomy, are also used to destroy nerves that transmit activating signals to sweat glands. |
To compete successfully in this market, we will have to provide an attractive alternative to these existing and other new therapies. Such competition could lead to reduced market share for sofpironium bromide and contribute to downward pressure on the pricing of sofpironium bromide, which could harm our business, financial condition, operating results and prospects.
Due to less stringent regulatory requirements in certain foreign countries, there are many more dermatological products and procedures available for use in those international markets than are approved for use in the United States. In certain international markets, there are also fewer limitations on the claims that our competitors can make about the effectiveness of their products and the manner in which they can market them. As a result, we expect to face more competition in these markets than in the United States.
We may in the future face generic competition for sofpironium bromide, which could expose us to litigation or adversely affect our business, financial condition, operating results and prospects.
Upon expiration of patent protection (including applicable extensions) in the United States (and any other countries where patent coverage exists) for sofpironium bromide, we could lose a significant portion of then-existing sales of sofpironium bromide in a short period of time from generic competition, which could expose us to litigation and would adversely affect our business, financial condition, operating results and prospects.
We have in the past relied, and expect to continue to rely, on third-party CROs and other third parties to conduct and oversee our sofpironium bromide clinical trials. If these third parties do not meet our requirements or otherwise conduct the trials as required or are unable to staff our trials, we may not be able to satisfy our contractual obligations or obtain regulatory approval for, or commercialize, sofpironium bromide.
We have in the past relied, and expect to continue to rely, on third-party contract research organizations (“CROs”) to conduct and oversee our sofpironium bromide clinical trials and other aspects of product development. We also rely on various medical institutions, clinical investigators and contract laboratories to conduct our trials in accordance with our clinical protocols and all applicable regulatory requirements, including the FDA’s regulations and good clinical practice (“GCP”) requirements, which are an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators and monitors, and state regulations governing the handling, storage, security and recordkeeping for drug and biologic products. These CROs and other third parties play a significant role in the conduct of these trials and the subsequent collection and analysis of data from the clinical trials. We rely heavily on these parties for the execution of our clinical trials and preclinical studies, and control only certain aspects of their activities. We and our CROs and other third-party contractors are required to comply with GCP and good laboratory practice (“GLP”) requirements, which are regulations and guidelines enforced by the FDA and comparable foreign regulatory authorities for sofpironium bromide. Regulatory authorities enforce these GCP and GLP requirements through periodic inspections of trial sponsors, principal investigators and trial sites. If we or any of these third parties fail to comply with applicable GCP and GLP requirements, or reveal noncompliance from an audit or inspection, the clinical data generated in our clinical trials may be deemed unreliable and the FDA or other regulatory authorities may require us to perform additional clinical trials before approving our or our partners’ marketing applications. We cannot assure that upon inspection by a given regulatory authority, such regulatory authority will determine that any of our clinical or preclinical trials comply with applicable GCP and GLP requirements. In addition, our clinical trials generally must be conducted with product produced under cGMP regulations.
12
Our failure to comply with these regulations and policies may require us to extend or repeat clinical trials, which would delay the regulatory approval process.
If any of our CROs or clinical trial sites terminate their involvement in one of our clinical trials for any reason, or are unable to continue to support us due to delay in implementation of the clinical trials due to the Bodor Complaint, we may not be able to enter into arrangements with alternative CROs or clinical trial sites, or do so on commercially reasonable terms, and in a satisfactory timeframe. In addition, if our relationship with clinical trial sites is terminated, we may experience the loss of follow-up information on patients enrolled in our ongoing clinical trials unless we are able to transfer the care of those patients to another qualified clinical trial site. In addition, principal investigators for our clinical trials may serve as scientific advisors or consultants to us from time to time and could receive cash or equity compensation in connection with such services. If these relationships and any related compensation result in perceived or actual conflicts of interest, the integrity of the data generated at the applicable clinical trial site may be questioned by the FDA.
We currently have limited marketing capabilities and no sales organization. If we are unable to establish sales and marketing capabilities on our own or through third parties, or are delayed in establishing these capabilities, we will be unable to successfully commercialize our product candidates, if approved, or generate product revenue.
We currently have limited marketing capabilities and no sales organization. To commercialize our product candidates, if approved, in the United States, Canada, the European Union, Latin America and other jurisdictions we seek to enter, we must build our marketing, sales, distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services, and we may not be successful in doing so. Although our employees have experience in the marketing, sale and distribution of pharmaceutical products, and business development activities involving external alliances, from prior employment at other companies, we as a company have no prior experience in the commercial launch, marketing, sale and distribution of pharmaceutical products, and there are significant risks involved in building and managing a sales organization, including our ability to hire, retain and incentivize qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel, and effectively manage a geographically dispersed sales and marketing team. Any failure or delay in the development of our internal sales, marketing, distribution and pricing/reimbursement/access capabilities would impact adversely the commercialization of these products.
To commercialize sofpironium bromide in Asia, we also intend to leverage the commercial infrastructure of our partner, Kaken, which will provide us with resources and expertise in certain areas that are greater than we could initially build ourselves. We may choose to collaborate with additional third parties in various countries that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales force and distribution systems. If we are unable to enter into such arrangements on acceptable terms or at all, we may not be able to successfully commercialize our product candidates, especially in other countries where we currently do not have a foreign legal presence. The inability to commercialize successfully our product candidates, either on our own or through collaborations with one or more third parties, would harm our business, financial condition, operating results and prospects.
Risks Related to Our Business
We currently have no products approved for sale, and we may never obtain regulatory approval to commercialize any of our product candidates.
The research, testing, manufacturing, safety surveillance, efficacy, quality assurance and control, recordkeeping, labeling, packaging, storage, approval, sale, marketing, distribution, import, export and reporting of safety and other post-market information related to our drug products are subject to extensive regulation by the FDA and other regulatory authorities in the United States and in foreign countries, and such regulations differ from country to country and frequently are revised.
Even after we or our partners achieve U.S. regulatory approval for a product candidate, if any, we or our partners will be subject to continued regulatory review and compliance obligations, including on how the product is commercialized. For example, with respect to our product candidates, the FDA may impose significant restrictions on the approved indicated use(s) for which the product may be marketed or on the conditions of approval. A product candidate’s approval may contain requirements for potentially costly post-approval studies and surveillance, including Phase 4 clinical trials, to monitor the safety and efficacy of the product or include in the approved label restrictions on the product and how it may be used or sold. We also will be subject to ongoing FDA obligations and continued regulatory review with respect to, among other things, the
13
manufacturing, processing, labeling, packaging, distribution, pharmacovigilance and adverse event reporting, storage, advertising, promotion and recordkeeping for our product candidates. These requirements include submissions of safety and other post-marketing information and reports, registration, continued compliance with cGMP requirements and with the FDA’s GCP requirements and GLP requirements, which are regulations and guidelines enforced by the FDA for all of our product candidates in clinical and preclinical development, and for any clinical trials that we conduct post-approval, as well as continued compliance with the FDA’s laws governing commercialization of the approved product, including but not limited to the FDA’s Office of Prescription Drug Promotion (“OPDP”) regulation of promotional activities, fraud and abuse, antikickback, product sampling, debarment, scientific speaker engagements and activities, formulary interactions as well as interactions with healthcare practitioners, including various conflict-of-interest reporting requirements for any healthcare practitioners we may use as consultants, and laws relating to the pricing of drug products, including federal “best price” regulations that if not met can prohibit the company from participating in federal reimbursement programs like Medicare or Medicaid. To the extent that a product candidate is approved for sale in other countries, we may be subject to similar or more onerous (i.e., prohibition on direct-to-consumer advertising that does not exist in the United States) restrictions and requirements imposed by laws and government regulators, and even private institutions, in those countries.
In addition, manufacturers of drug and biologic products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP regulations. If we or a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the manufacturing, processing, distribution or storage facility where, or processes by which, the product is made, a regulatory agency may impose restrictions on that product or us, including requesting that we initiate a product recall, or requiring notice to physicians or the public, withdrawal of the product from the market, or suspension of manufacturing.
If we, our partners, our product candidates or the manufacturing facilities for our product candidates fail to comply with applicable regulatory requirements, a regulatory agency may:
• | impose restrictions on the sale, marketing or manufacturing of the product, amend, suspend or withdraw product approvals or revoke necessary licenses; |
• | mandate modifications to or prohibit promotional and other product-specific materials or require us to provide corrective information to healthcare practitioners and other customers and/or patients, or in our advertising and promotion; |
• | require us or our partners to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions, penalties for noncompliance and, in extreme cases, require an independent compliance monitor to oversee our activities; |
• | issue warning letters, bring enforcement actions, initiate surprise inspections, issue show cause notices or untitled letters describing alleged violations, which may be publicly available; |
• | commence criminal investigations and prosecutions; |
• | debar certain healthcare professionals; |
• | exclude us from participating in or being eligible for government reimbursement and formulary inclusion; |
• | initiate audits, inspections, accounting and civil investigations or litigation; |
• | impose injunctions, suspensions or revocations of necessary approvals or other licenses; |
• | impose other civil or criminal penalties; |
• | suspend or cancel any ongoing clinical trials; |
• | place restrictions on the kind of promotional activities that can be done; |
14
• | delay or refuse to approve pending applications or supplements to approved applications filed by us or our potential partners; |
• | refuse to permit drugs or precursor chemicals to be imported or exported to or from the United States; |
• | suspend or impose restrictions on operations, including costly new manufacturing requirements; or |
• | seize or detain products or require us or our partners to initiate a product recall. |
The regulations, policies or guidance of the FDA and other applicable government agencies may change quickly, and new or additional statutes or government regulations may be enacted, including at the state and local levels, which can differ by geography and could prevent or delay regulatory approval of our product candidates or further restrict or regulate post-approval activities, including commercial efforts. We cannot predict the likelihood, nature or extent of adverse government regulations that may arise from future legislation or administrative action, either in the United States or abroad. If we are not able to achieve and maintain regulatory compliance, we may not be permitted to commercialize our product candidates, which would adversely affect our ability to generate revenue and achieve or maintain profitability.
We have sponsored or supported and may in the future sponsor or support clinical trials for our product candidates outside the United States, and the FDA and applicable foreign regulatory authorities may not accept data from such trials.
We have sponsored or supported and may in the future choose to sponsor or support one or more of our clinical trials outside of the United States. Although the FDA or applicable foreign regulatory authority may accept data from clinical trials conducted outside the United States or the applicable jurisdiction, acceptance of such study data by the FDA or applicable foreign regulatory authority may be subject to certain conditions or exclusion. Where data from foreign clinical trials are intended to serve as the basis for marketing approval in the United States, the FDA will not approve the application on the basis of foreign data alone unless such data are applicable to the U.S. population and U.S. medical practice; the studies were performed by clinical investigators of recognized competence; and the data are considered valid without the need for an on-site inspection by the FDA or, if the FDA considers such an inspection to be necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means. Many foreign regulatory bodies have similar requirements. In addition, such foreign studies would be subject to the applicable local laws of the foreign jurisdictions where the studies are conducted. There can be no assurance the FDA or applicable foreign regulatory authority will accept data from trials conducted outside of the United States or the applicable home country. If the FDA or applicable foreign regulatory authority does not accept such data, it would likely result in the need for additional trials, which would be costly and time-consuming and delay aspects of our business plan.
We may face product liability exposure, and if successful claims are brought against us, we may incur substantial liability if our insurance coverage for those claims is inadequate.
We face an inherent risk of product liability or similar causes of action as a result of the clinical testing of our product candidates and will face an even greater risk if we commercialize any products. This risk exists even if a product is approved for commercial sale by the FDA and is manufactured in facilities licensed and regulated by the FDA or an applicable foreign regulatory authority and notwithstanding that we comply with applicable laws on promotional activity. Our products and product candidates are designed to affect important bodily functions and processes. Any side effects, manufacturing defects, misuse or abuse associated with our product candidates could result in injury to a patient that may or may not be reversible or potentially even cause death. We cannot offer any assurance that we will not face product liability or other similar suits in the future or that we will be successful in defending them, nor can we assure that our insurance coverage will be sufficient to cover our liability under any such cases.
In addition, a liability claim may be brought against us even if our product candidates merely appear to have caused an injury. Product liability claims may be brought against us by consumers, healthcare providers, pharmaceutical companies or others selling or otherwise coming into contact with our product candidates, among others, and under some circumstances even government agencies. If we cannot successfully defend against product liability or similar claims, we will incur substantial liabilities, reputational harm and possibly injunctions and punitive actions. In addition, regardless of merit or eventual outcome, product liability claims may result in:
• | withdrawal or delay of recruitment or decreased enrollment rates of clinical trial participants; |
15
• | termination or increased government regulation of clinical trial sites or entire trial programs; |
• | the inability to commercialize our product candidates; |
• | decreased demand for our product candidates; |
• | impairment of our business reputation; |
• | product recall or withdrawal from the market or labeling, marketing or promotional restrictions; |
• | substantial costs of any related litigation or similar disputes; |
• | distraction of management’s attention and other resources from our primary business; |
• | significant delay in product launch; |
• | debarment of our clinical trial investigators or other related healthcare practitioners working with our company; |
• | substantial monetary awards to patients or other claimants against us that may not be covered by insurance; |
• | withdrawal of reimbursement or formulary inclusion; or |
• | loss of revenue. |
We have obtained product liability insurance coverage for our clinical trials. Large judgments have been awarded in class action or individual lawsuits based on drugs that had unanticipated side effects. Our insurance coverage may not be sufficient to cover all of our product liability-related expenses or losses and may not cover us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, restrictive and narrow, and, in the future, we may not be able to maintain adequate insurance coverage at a reasonable cost, or through self-insurance, in sufficient amounts or upon adequate terms to protect us against losses due to product liability or other similar legal actions. We will need to increase our product liability coverage if any of our product candidates receive regulatory approval, which will be costly, and we may be unable to obtain this increased product liability insurance on commercially reasonable terms or at all and for all geographies in which we wish to launch. A successful product liability claim or series of claims brought against us could, if judgments exceed our insurance coverage, decrease our cash, expose us to liability and harm our business, financial condition, operating results and prospects.
Our employees, independent contractors, principal investigators, other clinical trial staff, consultants, vendors, CROs and any partners with which we may collaborate may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
We are exposed to the risk that our employees, independent contractors, principal investigators, other clinical trial staff, consultants, vendors, CROs and any partners with which we may collaborate may engage in fraudulent or other illegal or unethical activity. Misconduct by these persons could include intentional, reckless, gross or negligent misconduct or unauthorized activity that violates: laws or regulations, including those laws requiring the reporting of true, complete and accurate information to the FDA or foreign regulatory authorities; product sampling; manufacturing standards; federal, state and foreign healthcare fraud and abuse laws and data privacy; anticorruption laws, anti-kickback and Medicare/Medicaid rules, debarment laws, promotional laws, securities laws, and/or laws that require the true, complete and accurate reporting of financial information or data, books and records. If any such or similar actions are instituted against us and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of civil, criminal and administrative and punitive penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal or state healthcare programs, debarments, contractual damages, reputational harm, diminished profits and future earnings, injunctions, and curtailment or cessation of our operations, any of which could expose us to liability and adversely affect our business, financial condition, operating results and prospects.
16
We may be subject to risks related to pre-approval promotion or off-label use, or unauthorized direct-to-consumer advertising of our product candidates.
The FDA strictly regulates the advertising and promotion of drug products, and drug products may only be marketed or promoted for their FDA-approved uses, consistent with the product’s approved labeling and to appropriate patient populations. Advertising and promotion of any product candidate that obtains approval in the United States will be heavily scrutinized by the FDA, the Department of Justice, the Office of Inspector General of the Department of Health and Human Services, state attorneys general, members of Congress, the public and others. Violations, including promotion of our products for unapproved or off-label uses, are subject to enforcement letters, inquiries and investigations, and civil, criminal and/or administrative sanctions by the FDA and other government agencies or tribunals and lawsuits by competitors, healthcare practitioners, consumers, investors or other plaintiffs. Additionally, advertising and promotion of any product candidate that obtains approval outside of the United States will be heavily scrutinized by relevant foreign regulatory authorities.
Even if we obtain regulatory approval for our product candidates, the FDA or comparable foreign regulatory authorities may require labeling changes or impose significant restrictions on a product’s indicated uses or marketing, or impose ongoing requirements for potentially costly post-approval studies or post-market surveillance.
In the United States, engaging in impermissible promotion of our product candidates for off-label uses, or engaging in pre-approval promotion of an unapproved drug candidate, also can subject us to false claims litigation under federal and state statutes, which can lead to civil, criminal and/or administrative penalties and fines and agreements, such as a corporate integrity agreement, that materially restrict the manner in which we promote or distribute our product candidates. If we do not lawfully promote our products once they have received regulatory approval, we may become subject to such litigation and, if we are not successful in defending against such actions, those actions could expose us to liability and could have a material adverse effect on our business, financial condition, operating results and prospects and even result in having an independent compliance monitor assigned to audit our ongoing operations at our cost for a lengthy period of time.
Other than sofpironium bromide, our product candidates are at the early stages of clinical and regulatory development.
We are evaluating the next clinical development steps for BBI-3000 and BBI‑6000, as each is in an early stage of clinical (prior to Phase 3) and preclinical development. The regulatory approval processes of the FDA and comparable foreign authorities are lengthy, time consuming, costly and inherently unpredictable, especially for early-stage product candidates. The time required to obtain approval for early stage product candidates from the FDA and comparable foreign authorities is unpredictable but typically takes many years, involves significant expenditures and depends upon numerous factors, including the substantial discretion of the regulatory authorities. In addition, approval policies, regulations, or the type and amount of clinical data necessary to gain approval may change during the course of a product candidate’s clinical development and may vary among jurisdictions. Our early stage product candidates will require substantial additional preclinical and clinical development before we will be able to submit an application to the FDA, if at all. Accordingly, we cannot assure you that we will be able to seek or obtain regulatory approval for any of our early stage product candidates.
Our clinical trials may fail to demonstrate the safety and efficacy of our other investigational agents BBI-3000 or BBI‑6000, or serious adverse or unacceptable side effects may be identified during their development, which could prevent or delay marketing approval and commercialization, increase our costs or necessitate the abandonment or limitation of the development of BBI-3000 or BBI-6000.
Before obtaining marketing approvals for the commercial sale of BBI-3000 and BBI-6000, we must demonstrate through lengthy, complex, uncertain and expensive preclinical testing and clinical trials that BBI-3000 and BBI-6000 are both safe and effective for use in each targeted indication, and failures can occur at any stage of testing. Clinical trials often fail to demonstrate safety and are associated with side effects or have characteristics that are unexpected. Based on the safety profile seen in clinical testing, we may need to abandon development or limit development to more narrow uses in which the side effects or other characteristics are less prevalent, less severe or more tolerable from a risk-benefit perspective. The FDA or an IRB also may require that we suspend, discontinue, or limit clinical trials based on safety information. Such findings could further result in regulatory authorities failing to provide marketing authorization for BBI-3000 or BBI-6000. Many drug candidates that initially showed promise in early-stage testing and which were efficacious have later been found to cause side effects that prevented further development of the drug candidate and, in extreme cases, the side effects were not seen until after the drug was marketed and exposed to large populations, causing regulators to remove the drug from the market post-approval.
17
We may choose not to continue developing or commercializing any of our early-stage product candidates at any time during development or after approval, which would reduce or eliminate our potential return on investment for those product candidates.
At any time, we may decide to discontinue the development of any of our early-stage product candidates for a variety of reasons, including the appearance of new technologies that make our product obsolete, competition from a competing product including entry of generics, supply chain considerations, intellectual property right impacts, ability to price or changes in or failure to comply with applicable regulatory requirements. If we terminate a program in which we have invested significant resources, we will not receive any return on our investment, and we will have missed the opportunity to have allocated those resources to potentially more productive uses. At this time, the company is preserving its resources to resolve the Bodor dispute.
Healthcare reform measures could hinder or prevent the commercial success of our product candidates.
The current presidential administration and certain members of the majority of the U.S. Congress have sought to repeal all or part of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, the “Affordable Care Act”), and implement a replacement program. For example, the so-called “individual mandate” was repealed as part of tax reform legislation adopted in December 2017, such that the shared responsibility payment for individuals who fail to maintain minimum essential coverage under section 5000A of the Internal Revenue Code was eliminated beginning in 2019. In addition, litigation may prevent some or all of the Affordable Care Act legislation from taking effect. For example, on December 14, 2018, the U.S. District Court for the Northern District of Texas held that the individual mandate is a critical and inseverable feature of the Affordable Care Act, and therefore, because it was repealed as part of the tax reform legislation, the remaining provisions of the Affordable Care Act are invalid as well. The impact of this ruling is stayed as it was appealed to the Fifth Circuit Court of Appeals. While the ruling will have no immediate effect, it is unclear how this decision, and subsequent appeals, if any, will impact the law. In 2020 and beyond, we may face additional uncertainties as a result of likely federal and administrative efforts to repeal, substantially modify or invalidate some or all of the provisions of the Affordable Care Act. There is no assurance that the Affordable Care Act, as amended in the future, will not adversely affect our business and financial results.
Additionally, in October 2018, the U.S. President proposed to lower Medicare Part B drug prices, in addition to contemplating other measures to lower or prescribe certain mandatory prescription drug prices or drug substitution policies. While these proposals have not yet been enacted, we expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our product candidates if approved or additional pricing pressures.
There are also calls to severely curtail or ban all direct-to-consumer advertising of pharmaceuticals, which would limit our ability to market our product candidates. The United States is already in a minority of jurisdictions that allow this kind of advertising and its removal could limit the potential reach of a marketing campaign.
We also may be subject to stricter healthcare laws, regulation and enforcement, and our failure to comply with those laws could expose us to liability or adversely affect our business, financial condition, operating results and prospects.
Certain federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to our business. We are subject to regulation by both the federal government and the states in which we or our partners conduct business. The healthcare laws and regulations that may affect our ability to operate include: the Federal Food, Drug and Cosmetic Act (FDCA), as amended; Title 21 of the Code of Federal Regulations Part 202 (21 CFR Part 202); the 21st Century Cures Act, the federal Anti-Kickback Statute; federal civil and criminal false claims laws and civil monetary penalty laws; the federal Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act; the Prescription Drug Marketing Act (for sampling of drug product among other things); the federal Best Price Act and Medicaid drug rebate program; the federal physician sunshine reporting requirements under the Affordable Care Act and state disclosure laws; the Foreign Corrupt Practices Act as it applies to activities both inside and outside of the United States; the new federal Right-to-Try legislation; and state law equivalents of many of the above federal laws.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of our business activities could be subject to challenge under one or more of such laws. In addition, recent
18
healthcare reform legislation has strengthened these laws. For example, the Affordable Care Act, among other things, amended the intent requirement of the federal Anti-Kickback Statute and certain criminal healthcare fraud statutes. A person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it. In addition, the Affordable Care Act provides that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act.
Achieving and sustaining compliance with these laws may prove costly. In addition, any action against us for violation of these laws, even if we successfully defend against it, could cause us to incur significant legal expenses and divert our management’s attention from the operation of our business and result in reputational damage. If our operations are found to be in violation of any of the laws described above or any other governmental laws or regulations that apply to us, we may be subject to penalties, including administrative, civil and criminal penalties, damages, including punitive damages, fines, disgorgement, the exclusion from participation in federal and state healthcare programs, individual imprisonment or corporate criminal liability, or the curtailment or restructuring of our operations, and injunctions, any of which could expose us to liability and could adversely affect our business, financial condition, operating results and prospects.
Subject to obtaining available financing, we intend to in-license and acquire product candidates and may engage in other strategic transactions, which could impact our liquidity, increase our expenses and present significant distractions to our management.
One of our strategies is to in-license and acquire product candidates and we may engage in other strategic transactions. Additional potential transactions that we may consider include a variety of different business arrangements, including mergers and acquisitions, spin-offs, strategic partnerships, joint ventures, co-marketing, co-promotion, distributorships, development and co-development, restructurings, divestitures, business combinations and investments on a global basis. Any such transaction(s) may require us to incur non-recurring or other charges, may increase our near- and long-term expenditures and may pose significant integration challenges or disrupt our management or business, which could adversely affect our operations and financial results. Accordingly, there can be no assurance that we will undertake or successfully complete any transactions of the nature described above, and any transaction that we do complete could expose us to liability and could harm our business, financial condition, operating results and prospects. We have no current plan, commitment or obligation to enter into any transaction described above other than ones to which we are already committed.
Our failure to in-license, acquire, develop and market successfully additional product candidates or approved products would impair our ability to grow our business.
We intend to in-license, acquire, develop and market additional products and product candidates. Because our internal research and development capabilities are limited, we may be dependent on pharmaceutical or other companies, investment groups or funds, academic or government scientists and other researchers to sell or license products or technology to us. The success of this strategy depends partly on our ability to identify and select promising pharmaceutical product candidates and products, negotiate licensing or acquisition agreements with their current owners, and finance these arrangements.
The process of proposing, negotiating and implementing a license or acquisition of a product candidate or approved product is lengthy and complex. Other companies, including some with substantially greater financial, marketing, sales, legal and other resources, may compete with us for the license or acquisition of product candidates and approved products. We have limited resources to identify and execute the acquisition or in-licensing of third-party products, businesses and technologies and integrate them into our current infrastructure. Moreover, we may devote resources to potential acquisitions or licensing opportunities that are never completed, or we may fail to realize the anticipated benefits of such efforts. We may not be able to acquire the rights to additional product candidates on terms that we find acceptable or at all.
Further, any product candidate that we acquire may require additional development efforts prior to commercial sale, including preclinical or clinical testing and approval by the FDA and applicable foreign regulatory authorities for the targeted use(s). All product candidates are prone to significant risks of failure typical of pharmaceutical product development, including the possibility that a product candidate will not be shown to be sufficiently safe and effective for approval by regulatory authorities. In addition, we cannot provide assurance that any approved products that we acquire will be manufactured or sold profitably, obtain reimbursement, be subject to patents and other intellectual property rights that provide any form of market or regulatory exclusivity, or achieve market acceptance.
19
Risks Related to Our Dependence on Third Parties
We expect to rely on our collaboration with third-party out-license partners for the successful development and commercialization of our product candidates.
We expect to rely upon the efforts of third-party out-license partners for the successful development and commercialization of our current and future product candidates. The clinical and commercial success of our product candidates may depend upon maintaining successful relationships with third-party out-license partners which are subject to a number of significant risks, including the following:
• | our partners’ ability to execute their responsibilities in a timely, cost-efficient and compliant manner; |
• | reduced control over delivery and manufacturing schedules; |
• | price increases and product reliability; |
• | manufacturing deviations from internal or regulatory specifications; |
• | quality incidents; |
• | the failure of partners to perform their obligations for technical, market, legal or other reasons; |
• | misappropriation of our current or future product candidates; and |
• | other risks in potentially meeting our current and future product commercialization schedule or satisfying the requirements of our end-users. |
We cannot assure you that we will be able to establish or maintain third-party out-license partner relationships in order to successfully develop and commercialize our product candidates.
We rely completely on third-party contractors to supply, manufacture and distribute clinical drug supplies for our product candidates, including certain sole-source suppliers and manufacturers; we intend to rely on third parties for commercial supply, manufacturing and distribution if any of our product candidates receive regulatory approval; and we expect to rely on third parties for supply, manufacturing and distribution of preclinical, clinical and commercial supplies of any future product candidates.
We do not currently have, nor do we plan to acquire, the infrastructure or internal capability to supply, store, manufacture or distribute preclinical, clinical or commercial quantities of drug substances or products. Additionally, we have not entered into a long-term commercial supply agreement to provide us with such drug substances or products. As a result, our ability to develop our product candidates is dependent, and our ability to supply our products commercially will depend, in part, on our ability to obtain the APIs and other substances and materials used in our product candidates successfully from third parties and to have finished products manufactured by third parties in accordance with regulatory requirements and in sufficient quantities for preclinical and clinical testing and commercialization. If we fail to develop and maintain supply and other technical relationships with these third parties, we may be unable to continue to develop or commercialize our products and product candidates.
We do not have direct control over whether our contract suppliers and manufacturers will maintain current pricing terms, be willing to continue supplying us with APIs and finished products or maintain adequate capacity and capabilities to serve our needs, including quality control, quality assurance and qualified personnel. We are dependent on our contract suppliers and manufacturers for day-to-day compliance with applicable laws and cGMPs for production of both APIs and finished products. If the safety or quality of any product or product candidate or component is compromised due to a failure to adhere to applicable laws or for other reasons, we may not be able to commercialize or obtain regulatory approval for the affected product or product candidate successfully, and we may be held liable for injuries sustained as a result.
In order to conduct larger or late-stage clinical trials for our product candidates and supply sufficient commercial quantities of the resulting drug product and its components, if that product candidate is approved for sale, our contract
20
manufacturers and suppliers will need to produce our drug substances and product candidates in larger quantities, more cost-effectively and, in certain cases, at higher yields than they currently achieve. If our third-party contractors are unable to scale up the manufacture of any of our product candidates successfully in sufficient quality and quantity and at commercially reasonable prices, or are shut down or put on clinical hold by government regulators, and we are unable to find one or more replacement suppliers or manufacturers capable of production at a substantially equivalent cost in substantially equivalent volumes and quality, and we are unable to transfer the processes successfully on a timely basis, the development of that product candidate and regulatory approval or commercial launch for any resulting products may be delayed, or there may be a shortage in supply, either of which could significantly harm our business, financial condition, operating results and prospects.
We expect to continue to depend on third-party contract suppliers and manufacturers for the foreseeable future. Our supply and manufacturing agreements, if any, do not guarantee that a contract supplier or manufacturer will provide services adequate for our needs. Additionally, any damage to or destruction of our third-party manufacturers’ or suppliers’ facilities or equipment, even by force majeure, may significantly impair our ability to have our products and product candidates manufactured on a timely basis. Our reliance on contract manufacturers and suppliers further exposes us to the possibility that they, or third parties with access to their facilities, will have access to and may misappropriate our trade secrets or other proprietary information. In addition, the manufacturing facilities of certain of our suppliers may be located outside of the United States. This may give rise to difficulties in importing our products or product candidates or their components into the United States or other countries.
Manufacturing and supply of the APIs and other substances and materials used in our product candidates and finished drug products is a complex and technically challenging undertaking, and there is potential for failure at many points in the manufacturing, testing, quality control and assurance and distribution supply chain, as well as the potential for latent defects after products have been manufactured and distributed.
Manufacturing and supply of APIs, other substances and materials and finished drug products is technically challenging. Changes beyond our direct control can impact the quality, volume, price and successful delivery of our products and product candidates and can impede, delay, limit or prevent the successful development and commercialization of our products and product candidates. Mistakes and mishandling are not uncommon despite reasonable best efforts and can affect successful production and supply. Some of these risks include but are not limited to:
• | failure of our manufacturers to follow cGMP or other legal requirements or mishandling of or adulterating product while in production or in preparation for transit; |
• | inability of our contract suppliers and manufacturers to efficiently and cost-effectively increase and maintain high yields and batch quality, consistency and stability; |
• | difficulty in establishing optimal drug delivery substances and techniques, production and storage methods and packaging and shipment processes; |
• | challenges in designing effective drug delivery substances and techniques especially in light of competitor options; |
• | transportation and import/export risk, particularly given the global nature of our supply chain; |
• | delays in analytical results or failure of analytical techniques that we depend on for quality control/assurance and release of a product; |
• | natural disasters, strikes and labor disputes, war and terrorism, financial distress, lack of raw material supply, issues with facilities and equipment or other forms of disruption to business operations of our contract manufacturers and suppliers; and |
• | latent defects that may become apparent after a product has been released and even sold and used and that may result in recall and destruction of the product. |
21
Any of these factors could result in delays or higher costs in connection with our clinical trials, regulatory submissions, required approvals or commercialization of our products, which could expose us to liability or harm our business, financial condition, operating results and prospects.
Risks Related to Our Financial Operations
We will need to raise substantial additional financing in the future to fund our operations, which may not be available to us on favorable terms or at all.
Pending successful resolution of the Bodor Complaint and obtaining substantial additional funding, we intend to conserve our resources. The advancement of the Phase 3 clinical trials for sofpironium bromide has been negatively impacted by the Bodor Complaint. As a result, we have taken, and expect to continue to take, actions to reduce our cash spend, including delaying the start of the clinical trials and/or staff reductions. In December 2019, an estimated loss contingency of $1.0 million was recorded for the Bodor Complaint and we will continue to evaluate the adequacy of this estimate as the matter develops. Nonetheless, we will require substantial additional funds to conduct the costly and time-consuming clinical trials necessary to pursue regulatory approval of each potential product candidate and to continue the development of sofpironium bromide in new indications or uses including commencing the Phase 3 clinical trials for sofpironium bromide. Our future capital requirements will depend upon a number of factors, including but not limited to: the number and timing of future product candidates in the pipeline; progress with and results from preclinical testing and clinical trials; the ability to manufacture sufficient drug supplies to complete preclinical and clinical trials; the costs involved in preparing, filing, acquiring, prosecuting, maintaining and enforcing patent and other intellectual property claims; compliance with our material contracts including the licensing agreement for sofpironium bromide and resolution of the Bodor Complaint; the time and costs involved in obtaining regulatory approvals and favorable reimbursement or formulary acceptance for such product candidates; and overall stock market conditions and trends. Raising additional capital may be costly or difficult to obtain and could significantly dilute stockholders’ ownership interests or inhibit our ability to achieve our business objectives. If we raise additional funds through public or private equity offerings, the terms of these securities may include liquidation or other preferences that adversely affect the rights of our common stockholders. Further, to the extent that we raise additional capital through the sale of common stock or securities convertible or exchangeable into common stock, our stockholders’ ownership interests in our company will be diluted. In addition, any debt financing may subject us to fixed payment obligations and covenants limiting or restricting our ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If we raise additional capital through marketing and distribution arrangements or other collaborations, strategic alliances or licensing arrangements with third parties, we may have to relinquish certain valuable intellectual property or other rights to our product candidates, technologies, future revenue streams or research programs or grant licenses on terms that may not be favorable to us in one or more countries.
As a result of the Bodor Complaint, Novaquest terminated its Funding Agreement with us and we have lost significant market capitalization. Our ability to raise the significant additional funds required to commence the Phase 3 clinical trials for sofpironium bromide is uncertain and is limited given our small market capitalization. Even if we were to obtain sufficient funding, there can be no assurance that it will be available on terms acceptable to us or our stockholders.
Our operating results and liquidity needs could be affected negatively by global market fluctuations and economic downturn.
Our operating results and liquidity could be affected negatively by global economic conditions generally, both in the United States and elsewhere around the world. The market for discretionary pharmaceutical products, medical devices and procedures may be particularly vulnerable to unfavorable economic conditions. Some patients may consider sofpironium bromide as discretionary, and if full reimbursement for the product is not available, demand for the product may be tied to the discretionary, out-of-pocket cash-spending levels of our targeted patient populations. Domestic and international equity and debt markets have experienced and may continue to experience heightened volatility and turmoil based on domestic and international economic conditions and concerns. In the event these economic conditions and concerns continue or worsen and the markets continue to remain volatile, or a bear market ensues in the U.S. stock market given the current bull market is the longest on record, our operating results and liquidity could be affected adversely by those factors in many ways, including weakening demand for sofpironium bromide, making it more difficult for us to raise funds if necessary, and our stock price may decline.
22
Our stock price has been and may continue to be highly volatile, and our common stock may continue to be illiquid.
The market price of our common stock following the Merger has been subject to significant fluctuations. The closing price of our common stock fluctuated from $4.69 per share as of September 3, 2019, the first trading date following the closing of the Merger, to $1.53 per share as of February 7, 2020. Market prices for securities of biotechnology and other life sciences companies historically have been particularly volatile subject even to large daily price swings. In addition, there has been limited liquidity in the trading market for our securities, which may adversely affect stockholders. Some of the factors that may cause the market price of our common stock to continue to fluctuate include, but are not limited to:
• | material developments in, or the conclusion of, any litigation to enforce or defend any intellectual property rights or defend against the intellectual property rights of others; |
• | the entry into, or termination of, or breach by us or our partners of material agreements, including key commercial partner or licensing agreements, including the License Agreement and the Kaken Agreement; |
• | our ability to obtain timely regulatory approvals for sofpironium bromide or future product candidates, and delays or failures to obtain such approvals; |
• | failure of sofpironium bromide, if approved, to achieve commercial success; |
• | issues in manufacturing sofpironium bromide or future product candidates; |
• | the results of current and any future clinical trials of sofpironium bromide; |
• | failure of other product candidates, if approved, to achieve commercial success; |
• | announcements of any dilutive equity financings; |
• | announcements by commercial partners or competitors of new commercial products, clinical progress or the lack thereof, significant contracts, commercial relationships or capital commitments; |
• | the introduction of technological innovations or new therapies or formulations that compete with sofpironium bromide; |
• | lack of commercial success of competitive products or products treating the same or similar indications; |
• | failure to elicit meaningful stock analyst coverage and downgrades of our stock by analysts; and |
• | the loss of key employees. |
Moreover, the stock markets in general have experienced substantial volatility in our industry that has often been unrelated to the operating performance of individual companies or a certain industry segment. These broad market fluctuations may also adversely affect the trading price of our common stock.
In the past, following periods of volatility in the market price of a company’s securities, shareholders have often instituted class action securities litigation against those companies. Such litigation, if instituted, could result in substantial costs and diversion of management attention and resources, which could significantly harm our profitability and reputation. In addition, such securities litigation often has ensued after a reverse merger or other merger and acquisition activity of the type we recently completed. Such litigation, if brought, could expose us to liability or impact negatively our business, financial condition, operating results and prospects.
Our operating results may fluctuate significantly, which makes our future operating results difficult to predict and could cause our operating results to fall below expectations.
23
Our operations to date have been limited primarily to researching and developing sofpironium bromide and undertaking preclinical studies and clinical trials of sofpironium bromide. We (and our partners) have not yet obtained regulatory approvals for sofpironium bromide in any country. Consequently, any predictions you or we make about our future success or viability may not be as accurate as they could be if we had a longer operating history or approved products on the market. Our revenue and profitability will depend on development funding, including obtaining the additional funds needed to commence the Phase 3 clinical trials for sofpironium bromide, and the achievement of development and clinical milestones under an agreement with Kaken, as well as any potential future collaboration and license agreements and sales of sofpironium bromide or future products, if approved, and our ability to maintain the related license as part of the Bodor Complaint. These up-front and milestone payments may vary significantly from period to period, and country to country, and any such variance could cause a significant fluctuation in our operating results from one period to the next. In addition, we will measure compensation cost for stock-based awards made to employees at the grant date of the award, based on the fair value of the award as determined by our board of directors and recognize the cost as an expense over the employee’s requisite service period. As the variables that we use as a basis for valuing these awards change over time, including our underlying stock price and stock price volatility, the magnitude of the expense that we must recognize may vary significantly. Furthermore, our operating results may fluctuate due to a variety of other factors, many of which are outside of our control and may be difficult to predict.
We incur costs and demands upon management as a result of complying with the laws and regulations affecting public companies.
We incur significant legal, accounting and other expenses that Brickell did not incur as a private company prior to the Merger and operating as a public company, including costs associated with public company reporting and other SEC requirements. We also incur costs associated with newly applicable corporate governance requirements, including requirements under the Sarbanes-Oxley Act, as well as new rules implemented by the SEC and The Nasdaq Stock Market LLC. These rules and regulations are expected to increase our legal and financial compliance costs and to make some activities more time-consuming and costly. Our executive officers and other personnel will need to devote substantial time to gaining expertise regarding operations as a public company and compliance with applicable laws and regulations. These rules and regulations may also make it expensive for us to operate our business.
We are a “smaller reporting company” and the reduced disclosure and governance requirements applicable to smaller reporting companies may make our common stock less attractive to some investors.
We qualify as a “smaller reporting company” under Rule 12b-2 of the Exchange Act. As a smaller reporting company, we are entitled to rely on certain exemptions and reduced disclosure requirements, such as simplified executive compensation disclosures and reduced financial statement disclosure requirements, in our SEC filings. These exemptions and decreased disclosures in our SEC filings due to our status as a smaller reporting company may make it harder for investors to analyze our results of operations and financial prospects. We cannot predict if investors will find our common stock less attractive because we rely on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock and our common stock price may be more volatile. We will remain a “smaller reporting company” under Item 10(f)(1) of SEC Regulation S-K as long as we maintain a public float as defined by that regulation of less than $250 million; or we have less than $100 million in annual revenues and (i) either no public float, or (ii) a public float of less than $700 million.
Provisions of Delaware law and our amended certificate of incorporation and amended and restated bylaws may discourage another company from acquiring us and may prevent attempts by our stockholders to replace or remove our current management.
Provisions of Delaware law and our amended certificate of incorporation and amended and restated bylaws may discourage, delay or prevent a merger or acquisition that our stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace or remove our board of directors. These provisions include, but are not limited to:
• | authorizing the issuance of “blank check” preferred stock without any need for action by stockholders; |
• | providing for a classified board of directors with staggered terms; |
24
• | requiring supermajority stockholder voting to effect certain amendments to our current certificate of incorporation and bylaws; |
• | eliminating the ability of stockholders to call special meetings of stockholders; and |
• | establishing advance notice requirements for nominations for election to our board of directors or for proposing matters that can be acted on by stockholders at stockholder meetings. |
Although we believe these provisions collectively provide for an opportunity to receive higher bids by requiring potential acquirers to negotiate with our board of directors, they would apply even if an offer may be considered beneficial by some stockholders. In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it difficult for stockholders to replace members of our board of directors, which is responsible for appointing the members of our management.
If the holders of our company’s stock options and warrants exercise their rights to purchase our common stock, the ownership of our stockholders will be diluted.
As of February 7, 2020, we have (i) warrants issued and outstanding to purchase one share of our common stock at an exercise price of $0.07 per share, 490,683 shares of our common stock at an exercise price of $10.36 per share and 9,005 shares of our common stock at an exercise price of $33.31 per share; and (ii) we have 1,793,602 options issued and outstanding to purchase our common stock at a weighted average exercise price of $13.00 per share. If the holders of our outstanding stock options and warrants exercise their rights to acquire our common stock, the percentage ownership of our stockholders existing prior to the exercise of such rights will be diluted.
We do not anticipate paying any dividends in the foreseeable future.
Our current expectation is that we will retain our future earnings to fund the development and growth of our business. As a result, capital appreciation, if any, of our shares will be your sole source of gain, if any, for the foreseeable future.
If we fail to attract and retain management and other key personnel and directors, we may be unable to continue to successfully develop or commercialize our product candidates or otherwise implement our business plan.
Our ability to compete in the highly competitive pharmaceuticals industry depends on our ability to attract and retain highly qualified managerial, scientific, medical, legal, sales and marketing and other personnel, and directors of our board of directors. We are highly dependent on our management and scientific personnel and our directors. The loss of the services of any of these individuals could impede, delay or prevent the successful development of our product pipeline, completion of our planned clinical trials, commercialization of our product candidates or in-licensing or acquisition of new assets and could impact negatively our ability to implement successfully our business plan and in a way that complies with all applicable laws. If we lose the services of any of these individuals, we might not be able to find suitable replacements on a timely basis or at all, and our business could be harmed as a result. We might not be able to attract or retain qualified management and other key personnel or directors in the future due to the intense competition for qualified individuals among biotechnology, pharmaceutical and other businesses.
Our ability to use our net operating loss carryforwards to offset future taxable income may be subject to certain limitations.
As of December 31, 2018, we had approximately $36.5 million of federal and $30.9 million of state operating loss carryforwards available to offset future taxable income, which expire in varying amounts beginning in 2030 for federal and state purposes if unused. It is possible that we will not generate taxable income in time to use these loss carryforwards before their expiration. Our net operating loss carryforwards may also be subject to limitation as a result of prior shifts in equity ownership in connection with the Merger. In addition, we may experience ownership changes in the future as a result of offerings of our stock or subsequent shifts in our stock ownership, some of which are outside of our control. In that case, the ability to use net operating loss carryforwards to offset future taxable income will be limited following any such ownership change.
25
We may be adversely affected by natural disasters and other catastrophic events and by man-made problems such as war or terrorism or labor disruptions that could disrupt our business operations, and our business continuity and disaster recovery plans may not adequately protect us from a serious disaster.
Our corporate office is located in Boulder, Colorado, near a major flood and blizzard zone. If a disaster, power outage, computer hacking, or other event occurred that prevented us from using all or a significant portion of our office, that damaged critical infrastructure (such as enterprise financial systems, IT systems, manufacturing resource planning or enterprise quality systems), or that otherwise disrupted operations, it may be difficult or, in certain cases, impossible for us to continue our business for a substantial period of time. Our contract manufacturers’ and suppliers’ facilities are located in multiple locations where other natural disasters or similar events, such as tornadoes, earthquakes, storms, fires, explosions or large-scale accidents or power outages, or IT threats, could severely disrupt our operations, could expose us to liability and could have a material adverse effect on our business, financial condition, operating results and prospects. In addition, acts of terrorism and other geo-political unrest or labor unrest could cause disruptions in our business or the businesses of our partners, manufacturers or the economy as a whole. All of the aforementioned risks may be further increased if we do not implement a disaster recovery plan or our partners’ or manufacturers’ disaster recovery plans prove to be inadequate. To the extent that any of the above should result in delays in the regulatory approval, manufacture, distribution or commercialization of sofpironium bromide, this could expose us to liability, and our business, financial condition, operating results and prospects would suffer.
Our business and operations would suffer in the event of system failures, cyber-attacks or a deficiency in our cyber-security.
Despite the implementation of security measures, our internal computer systems and those of our current and future CROs and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, computer hacking or breaches, natural disasters, terrorism, war, labor unrest, and telecommunication and electrical failures. The risk of a security breach or disruption, particularly through cyber-attacks or cyber-intrusion, including by computer hackers, foreign governments, and cyber-terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. While we have not experienced any such material system failure, accident or security breach to date, if such an event were to occur and cause interruptions in our operations, it could result in a material disruption of our development programs and our business operations. In addition, since we sponsor clinical trials, any breach that compromises patient data and identities causing a breach of privacy could generate significant reputational damage and legal liabilities and costs to recover and repair, including affecting trust in us to recruit for future clinical trials. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in our regulatory approval efforts and significantly increase our costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, our data or applications or inappropriate disclosure of confidential or proprietary information, we could incur liability and the further development and commercialization of our products and product candidates could be delayed.
Risks Related to Our Intellectual Property
We may not be able to obtain, maintain or enforce global patent rights or other intellectual property rights that cover sofpironium bromide and related technologies that are of sufficient breadth.
Our success with respect to sofpironium bromide will depend, in part, on our ability to protect patent and other intellectual property protections in both the United States and other countries, to preserve our trade secrets and to prevent third parties from infringing on our proprietary rights. Our ability to prevent unauthorized or infringing use of sofpironium bromide by third parties depends in substantial part on our ability to leverage valid and enforceable patents and other intellectual property rights around the world.
The patent application process, also known as patent prosecution, is expensive and time-consuming, and we and our current or future licensors and licensees may not be able to prepare, file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner in all the countries that may be desirable. It is also possible that we or our current licensors and licensees, or any future licensors or licensees, will fail to identify patentable aspects of inventions made in the course of development and commercialization activities before it is too late to obtain patent protection by others on them. Therefore, these and any of our patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of our business. Moreover, our competitors independently may develop equivalent knowledge, methods and know-how or discover workarounds to our patents that would not constitute infringement. Any of these
26
outcomes could impair our ability to enforce the exclusivity of our patents effectively, which may have an adverse impact on our business, financial condition, operating results and prospects.
Due to constantly shifting global legal standards relating to patentability, validity, enforceability and claim scope of patents covering pharmaceutical inventions, our ability to protect patents in any jurisdiction is uncertain and involves complex legal and factual questions especially across countries. Accordingly, rights under any applicable patents that apply to us may not cover our product candidates or may not provide us with sufficient protection for our product candidates to afford a sustainable commercial advantage against competitive products or processes, including those from branded, generic and over-the-counter pharmaceutical companies. In addition, we cannot guarantee that any patents or other intellectual property rights will issue from any pending or future patent or other similar applications related to us. Even if patents or other intellectual property rights have issued or will issue, we cannot guarantee that the claims of these patents and other rights are or will be held valid or enforceable by the courts or other legal authorities, through injunction or otherwise, or will provide us with any significant protection against competitive products or otherwise be commercially valuable to us in every country of commercial significance that we may target, or that a legislative or executive branch of government may alter the rights and enforceability thereof at any time.
Competitors in the field of dermatologic therapeutics have created a substantial amount of prior art, including scientific publications, abstracts, posters, presentations, patents and patent applications and other public disclosures including on the Internet and various social media. Our ability to protect valid and enforceable patents and other intellectual property rights depends on whether the differences between our proprietary technology and the prior art allow our technology to be patentable over the prior art. We do not have outstanding issued patents covering all of the recent developments in our technology and are unsure of the patent protection that we will be successful in securing, if any. Even if the patents do issue successfully, third parties may design around or challenge the validity, enforceability or scope of such issued patents or any other issued patents or intellectual property that apply to us, which may result in such patents and/or other intellectual property being narrowed, invalidated or held unenforceable. If the breadth or strength of protection provided by the patents and other intellectual property we hold or pursue with respect to our product candidates is challenged, regardless of our future success, it could dissuade companies from collaborating with us to develop, or threaten our ability to commercialize or finance, our product candidates.
The laws of some foreign jurisdictions do not provide intellectual property rights to the same extent or duration as in the United States, and many companies have encountered significant difficulties in acquiring, maintaining, protecting, defending and especially enforcing such rights in foreign jurisdictions. If we encounter such difficulties in protecting, or are otherwise precluded from effectively protecting, our intellectual property in foreign jurisdictions, our business prospects could be substantially harmed, especially internationally.
Patents have a limited lifespan. In the United States, the natural expiration of a patent is generally 20 years after it is filed, with patent term extensions granted in certain instances to compensate for part of the period in which the drug was under development and could not be commercialized while under the patent. Without patent protection for sofpironium bromide, we may be open to competition from generic versions of sofpironium bromide. The issued U.S. patents relating to sofpironium bromide run through 2031, including expected extensions just described. Other patent rights we are seeking in the United States would provide expected coverage through 2040, but only in the event of a grant of such rights.
Proprietary trade secrets and unpatented know-how are also very important to our business. Although we have taken steps to protect our trade secrets and unpatented know-how by entering into confidentiality agreements with third parties and intellectual property protection agreements with officers, directors, employees, and certain consultants and advisors, there can be no assurance that binding agreements will not be breached or enforced by courts or other legal authorities, that we would have adequate remedies for any breach, including injunctive and other equitable relief, or that our trade secrets and unpatented know-how will not otherwise become known, be inadvertently disclosed by us or our agents and representatives, or be independently discovered by our competitors. If trade secrets are independently discovered, we would not be able to prevent their use and if we and our agents or representatives inadvertently disclose trade secrets and/or unpatented know-how, we may not be allowed to retrieve the inadvertently disclosed trade secret and/or unpatented know-how and maintain the exclusivity we previously enjoyed.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on our product candidates does not guarantee exclusivity. The requirements for patentability differ in certain countries, particularly developing countries, and can change over time in the
27
same country. In addition, the laws of some other countries do not protect intellectual property rights to the same extent as laws in the United States, especially when it comes to granting use and other kinds of patents and what kind of enforcement rights will be allowed, especially injunctive relief in a civil infringement proceeding. Consequently, we may not be able to prevent third parties from practicing our inventions in countries outside the United States and even in launching an identical version of our product notwithstanding us having a valid patent or other intellectual property rights in that country. Competitors may use our technologies in jurisdictions where we or our licensors have not obtained patent or other protections to develop their own products, or produce copy products, and, further, may export otherwise infringing products to territories where we have patent and other protections but enforcement against infringing activities is inadequate or where we have no patents or other intellectual property rights. These products may compete with our products, and our patents or other intellectual property rights may not be effective or sufficient to prevent them from commercialization or other uses.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly in developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to pharmaceuticals, and the judicial and government systems are often corrupt, apathetic or ineffective, which could make it difficult for us to stop the infringement of our patents or marketing of competing products in violation of our intellectual property rights generally. Proceedings to enforce our intellectual property rights in foreign jurisdictions could result in substantial costs and divert our efforts and attention from other aspects of our business, could put our global patents and other rights at risk of being invalidated or interpreted narrowly and our global patent applications at risk of not issuing, and could provoke third parties to assert claims against us. We may not prevail in any lawsuit that we initiate or infringement action brought against us, and the damages or other remedies awarded, if any, may not be commercially meaningful when we are the plaintiff. When we are the defendant, we may be required to post large bonds to stay in the market while we defend ourselves from an infringement action.
In addition, certain countries in Europe and certain developing countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties, especially if the patent owner does not enforce or use its patents over a protracted period of time. In some cases, the courts will force compulsory licenses on the patent holder even when finding the patentholder’s patents are valid if the court believes it is in the best interests of the country to have widespread access to an essential product covered by the patent. Further, there is no guarantee that any country will not adopt or impose compulsory licensing in the future. In these situations, the royalty the court requires to be paid by the licenseholder receiving the compulsory license may not be calculated at fair market value and can be inconsequential, thereby disaffecting the patentholder’s business. In these countries, we may have limited remedies if our patents are infringed or if we are compelled to grant a license to our patents to a third party, which could also materially diminish the value of those patents. This would limit our potential revenue opportunities. Accordingly, our efforts to enforce our intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that we own or license, especially in comparison to what we enjoy from enforcing our intellectual property rights in the United States. Finally, our ability to protect and enforce our intellectual property rights may be adversely affected by unforeseen changes in both U.S. and foreign intellectual property laws, or changes to the policies in various government agencies in these countries, including but not limited to the patent office issuing patents and the health agency issuing pharmaceutical product approvals. For example, in Brazil, pharmaceutical patents require prior initial approval of the Brazilian health agency (ANVISA). Finally, many countries have large backlogs in patent prosecution, and in some countries in Latin America it can take years, even decades, just to get a pharmaceutical patent application reviewed notwithstanding the merits of the application.
Obtaining and maintaining our patent protection depends on compliance with various procedural, document submission, fee payment, and other requirements imposed by governmental patent and similar agencies, and our patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance and annuity fees on any issued patent are due to be paid to the U.S. Patent and Trademark Office (“USPTO”) and foreign patent agencies in several stages over the lifetime of a patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can, in many cases, be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction just for failure to know about and/or timely pay such fee. Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non-payment of fees in prescribed time periods, and failure to properly legalize and submit formal documents in the format and style the country requires. If we or our licensors fail to maintain the patents and patent applications covering our
28
product candidates for any reason, our competitors might be able to otherwise enter the market, which would have an adverse effect on our business, financial condition, operating results and prospects.
In addition, countries continue to increase the fees that are charged to acquire, maintain and enforce patents and other intellectual property rights, which may become prohibitive to initiate or continue paying in certain circumstances.
If we fail to comply with our obligations under our intellectual property license agreements, we could lose license rights that are important to our business. Additionally, these agreements may be subject to disagreement over contract interpretation, which could narrow the scope of our rights to the relevant intellectual property or technology, or increase our financial or other obligations to our licensors.
We have entered into in-license arrangements with respect to certain of our product candidates. These license agreements impose various diligence, milestone, royalty, insurance, reporting and other obligations on us. If we fail to comply with these obligations, the respective licensors may have the right to terminate or modify the license, or trigger other more disadvantageous contract clauses, in which event we may not be able to finance, develop or market the affected product candidate. The loss of such rights could expose us to liability and could materially adversely affect our business, financial condition, operating results and prospects.
Our commercial success depends on our ability to develop, manufacture, market and sell our product candidates and use our proprietary technologies without infringing the proprietary rights of third parties and do this in one or more countries. We cannot assure that marketing and selling such product candidates and using such technologies will not infringe existing or future patents. Numerous U.S.- and foreign-issued patents and pending patent applications owned by third parties exist in the fields relating to our product candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that others may assert that our product candidates, technologies or methods of delivery or use(s) infringe their patent or other intellectual property rights. Moreover, it is not always clear to industry participants, including us, which patents and other intellectual property rights cover various drugs, biologics, drug delivery systems and formulations, manufacturing processes, or their methods of use, and which of these patents may be valid and enforceable. Thus, because of the large number of patents issued and patent applications filed in our fields across many countries, there may be a risk that third parties may allege they have patent or other rights encompassing our product candidates, technologies or methods.
In addition, there may be issued patents of third parties that are infringed or are alleged to be infringed by our product candidates or proprietary technologies notwithstanding the patents we may possess. Because some patent applications in the United States and other countries may be maintained in confidence until the patents are issued, because patent applications in the United States and many foreign jurisdictions are typically not published until 18 months or some other time after filing, and because publications in the scientific literature or other public disclosures often lag behind actual discoveries, we cannot be certain that others have not filed patent applications for technology covered by our patents or our pending applications. Our competitors may have filed, and may in the future file, patent applications covering our product candidates or technology similar to our technology. Any such patent application may have priority over our patent applications or patents, which could further require us to obtain rights to issued patents covering such technologies, which may mean paying significant licensing fees or royalties, or the like. If another party has filed a U.S. patent application on inventions similar to ours, we or the licensor, may have to participate in the United States in an interference proceeding to determine priority of invention.
We may be exposed to, or threatened with, future litigation by third parties having patent or other intellectual property rights alleging that our product candidates or proprietary technologies infringe such third parties’ intellectual property rights, including litigation resulting from filing in the United States under Paragraph IV of the Hatch-Waxman Act or other countries’ laws similar to the Hatch-Waxman Act. These lawsuits could claim that there are existing patent rights for such drug, and this type of litigation can be costly and could adversely affect our operating results and divert the attention of managerial and technical personnel, even if we do not infringe such patents or the patents asserted against us are ultimately established as invalid. There is a risk that a court or other legal authority would decide that we are infringing the third party’s patents and would order us to stop the activities covered by the patents. In addition, there is a risk that a court or other legal authority will order us to pay the other party significant damages for having violated the other party’s patents or intellectual property rights.
Because we rely on certain third-party licensors and partners and will continue to do so in the future, around the world, if one of our licensors or partners is sued for infringing a third party’s intellectual property rights, this could expose us
29
to liability and our business, financial condition, operating results and prospects could suffer in the same manner as if we were sued directly. In addition to facing litigation risks, we have agreed to indemnify certain third-party licensors and partners against claims of infringement caused by our proprietary technologies, and we have entered or may enter into cost-sharing agreements with some of our licensors and partners that could require us to pay some of the costs of patent or other intellectual property rights litigation brought against those third parties whether or not the alleged infringement is caused by our proprietary technologies. In certain instances, these cost-sharing agreements could also require us to assume greater responsibility for infringement damages than would be assumed just on the basis of our technology.
The occurrence of any of the foregoing could expose us to liability or adversely affect our business, financial condition, operating results and prospects at any time.
We may be subject to claims that our officers, directors, employees, consultants or independent contractors have wrongfully used or disclosed to us alleged trade secrets or other confidential and proprietary information of their former employers or their former or current customers.
As is common in the biotechnology and pharmaceutical industries, certain of our employees were formerly employed by other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Moreover, we engage the services of consultants to assist us in the development of our products and product candidates, many of whom were previously employed at, or may have previously been or are currently providing consulting services to, other biotechnology or pharmaceutical companies, including our competitors or potential competitors. We may be subject to claims that these employees and consultants or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary confidential information of their former employers or their former or current customers. Although we have no knowledge of any such claims being alleged to date, if such claims were to arise, litigation may be necessary to defend against any such claims. Even if we are successful in defending against any such claims, any litigation like this could be protracted, expensive, a distraction to our management team, not viewed favorably by investors and other third parties, and may potentially result in an unfavorable outcome.
30
BUSINESS
Overview
We are a clinical-stage pharmaceutical company focused on the development of innovative and differentiated prescription therapeutics for the treatment of debilitating skin diseases. Our pipeline consists of potential novel therapeutics for hyperhidrosis, cutaneous T-cell lymphoma, psoriasis, and other prevalent dermatological conditions. Our executive management team and board of directors bring extensive experience in product development and global commercialization, having served in leadership roles at large global pharmaceutical companies and biotechs that have developed and/or launched successful products, including several that were first-in-class and/or achieved iconic status, such as Cialis®, Taltz®, Gemzar®, Prozac®, Cymbalta® and Juvederm®.
Our pivotal Phase 3-ready clinical-stage product candidate, sofpironium bromide, is a proprietary new molecular entity. It belongs to a class of medications called anticholinergics. Anticholinergics block the action of acetylcholine, a chemical that transmits signals within the nervous system that are responsible for a range of bodily functions, including activation of the sweat glands. Sofpironium bromide was retrometabolically designed. Retrometabolic drugs are designed to exert their action topically and are potentially rapidly metabolized once absorbed into the blood. This proposed mechanism of action may allow for highly effective doses to be used while limiting systemic side effects. We are developing sofpironium bromide as a potential best-in-class, self-administered, once-daily, topical therapy for the treatment of primary axillary hyperhidrosis. Hyperhidrosis is a life-altering condition of sweating beyond what is physiologically required to maintain normal thermal regulation. It is believed to be caused by an overactive cholinergic response of the sweat glands and affects an estimated 15.3 million, or 4.8%, of the U.S. population. According to a 2016 update on the prevalence and severity of hyperhidrosis in the United States by Doolittle et al., axillary (underarm) hyperhidrosis, which is the targeted first potential indication for sofpironium bromide, is the most common occurrence of hyperhidrosis, affecting approximately 65% of patients in the United States or an estimated 10 million individuals.
We and our development partner in Asia, Kaken, have conducted 19 clinical trials of sofpironium bromide gel that encompass over 1,200 subjects in the United States and Japan. These trials evaluated the potential safety, tolerability, pharmacokinetics, and efficacy of sofpironium bromide gel in adult and pediatric primary axillary hyperhidrosis patients and healthy adult subjects. Under the Kaken Agreement, in exchange for paying us an upfront, nonrefundable payment, we granted Kaken the exclusive right to develop, manufacture and commercialize sofpironium bromide in Japan and certain other Asian countries. In March 2019, Kaken completed a Phase 3 trial in patients with primary axillary hyperhidrosis in Japan, achieving statistical significance on all primary and secondary endpoints. In January 2020, we announced that Kaken submitted a new drug application for approval of manufacturing and marketing for sofpironium bromide in Japan for primary axillary hyperhidrosis.
Based on the positive results of the clinical trials for sofpironium bromide to date, we intend to initiate two pivotal Phase 3 clinical trials in up to 350 subjects per trial with primary axillary hyperhidrosis in the United States, subject to obtaining substantial additional funding and the successful resolution of our dispute with the Bodor Plaintiffs. Assuming the results of the Phase 3 clinical trials are favorable, we plan thereafter to submit an NDA to the FDA for the treatment of hyperhidrosis by sofpironium bromide.
On October 23, 2019, Bodor notified us of its purported termination of the License Agreement. In connection with this attempt, on October 23, 2019, the Bodor Plaintiffs filed the Bodor Complaint against us in the United States District Court for the Southern District of Florida. The Bodor Complaint alleges damages incurred by the Bodor Plaintiffs in connection with our alleged material breach of the License Agreement and requested a declaratory judgment that the attempted termination of the License Agreement by Bodor be permitted. As required by the License Agreement, we initiated an arbitration proceeding and then subsequently a mediation with the Bodor Plaintiffs to resolve the dispute. In our arbitration demand, we countersued the Bodor Plaintiffs for damages caused by them to us, both under contract and due to their tortious interference with our business relations. We asked the district court to dismiss or stay the Bodor Complaint. The judge administratively closed the case pending arbitration. We believe that the claims and assertions made by the Bodor Plaintiffs are in substance without merit. Following an unsuccessful mediation, we and the Bodor Plaintiffs commenced arbitration. Pending successful resolution of this dispute and obtaining substantial additional funding, we intend to conserve our resources. The advancement of the Phase 3 clinical trials for sofpironium bromide have been negatively impacted by these developments and will require substantial additional funds. We have taken, and expect to continue to take, actions to reduce our cash spend, including delaying the start of the clinical trials and/or staff reductions.
31
Our second product candidate, BBI-3000, is a selective, potentially highly tolerable and potent novel RXR agonist that we are evaluating for the development in CTCL as a potential oral treatment. Retinoids are derivatives of vitamin A that play a pivotal role in a diverse group of biologic processes including, but not limited to, cellular proliferation, differentiation, apoptosis, and development. The biological activity and tolerability of retinoids depends in part on the binding availability to retinoic acid and RXR receptors. There are several topical and oral retinoids currently on the market that have shown efficacy in the treatment of several skin conditions, such as CTCL (e.g., bexarotene/Targretin®), acne and psoriasis (e.g., tazarotene, adapalene and tretinoin). BBI-3000 has been well tolerated in two Phase 1 studies (a single dose study and a multiple dose study) conducted by the NCI in healthy volunteers. There is an ongoing Phase 1b trial being conducted by the NCI to assess the biological effect of BBI-3000 on early stage breast cancer.
Our third product candidate, BBI-6000, is a novel RORg inhibitor that we are developing for the topical treatment of mild-to-moderate psoriasis. RORg inhibition targets the pathway of IL-17 that has been implicated in the pathogenesis of psoriasis. Monoclonal antibodies targeting IL‑17 have recently shown significant efficacy in the treatment of psoriasis, and we are planning to develop BBI‑6000 as a topically applied, potent and selective small-molecule therapeutic targeting this pathway. BBI-6000 is currently in the preclinical stages of development.
Merger of Brickell Biotech, Inc. and Vical Incorporated
On August 31, 2019, the Delaware corporation formerly known as “Vical Incorporated”, completed a reverse merger transaction in accordance with the terms and conditions of the Merger Agreement, by and among Vical, Brickell and Merger Sub, pursuant to which the Merger Sub merged with and into Brickell, with Brickell surviving the merger as a wholly-owned subsidiary of Vical. Additionally, on August 31, 2019, immediately after the completion of the Merger, the Company changed its name from “Vical Incorporated” to “Brickell Biotech, Inc.” On August 31, 2019, in connection with, and prior to the consummation of the Merger, Vical effected a reverse stock split of its common stock, par value $0.01 per share, at a ratio of 1-for-7.
Concurrent with the execution of the Merger Agreement, we entered into the Funding Agreement with NovaQuest pursuant to which NovaQuest committed partially to fund our expenses relating to certain product development activities. As a result of the dispute with the Bodor Plaintiffs, on November 25, 2019, Brickell Subsidiary, Inc., our wholly-owned subsidiary, and NovaQuest entered into the Settlement and Termination Agreement effectively terminating the Funding Agreement. NovaQuest agreed to cancel and surrender the warrant it previously received in connection with the Funding Agreement, and we repaid NovaQuest the $5.6 million advance previously made by NovaQuest in addition to accrued interest. Subject to the mutual indemnity included in the Settlement and Termination Agreement, NovaQuest agreed to waive any and all of our further obligations (including any and all future milestone payments and royalties owed to NovaQuest) and each party agreed to release any and all claims against the other party in respect of the Funding Agreement.
Our common stock trades on The Nasdaq Capital Market under the symbol “BBI.” Our principal executive offices are located at 5777 Central Avenue, Suite 102, Boulder, Colorado 80301, our telephone number is (720) 505-4755 and our corporate website address is www.brickellbio.com. Our website and the information contained on or accessible through our website are not part of this document. We have included our website address in this prospectus solely as an inactive textual reference.
32
Product Candidates
Our current portfolio of product candidates, all of which are new molecular entities accompanied with certain intellectual property rights, are summarized in the following chart for the U.S. market:
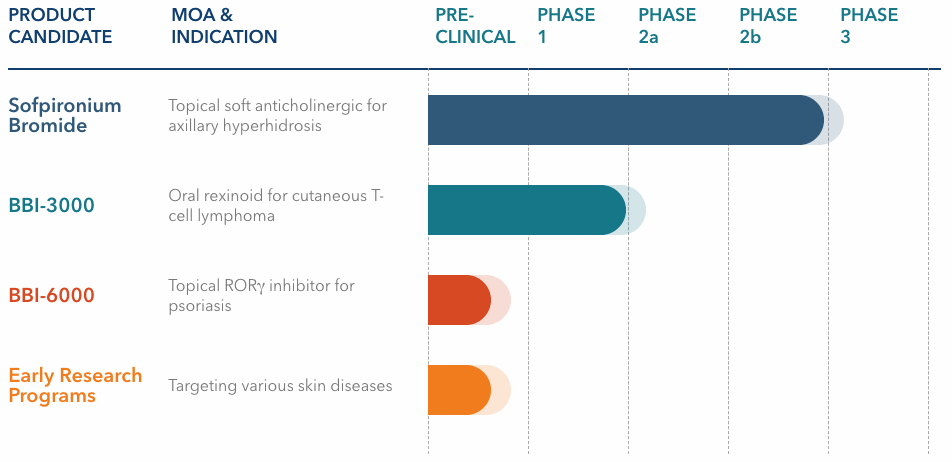
Hyperhidrosis
Hyperhidrosis is a debilitating life-altering skin disorder of chronic excessive sweating beyond what is necessary for thermoregulation of the body. Current estimates show that primary hyperhidrosis (excessive sweating without an alternative origin) affects approximately 4.8% of the U.S. population, or roughly 15.3 million people, with the prevalence highest (8.8%) among the U.S. population ages 18–39. Of these individuals, 70% report severe excessive sweating that they cannot control or shut off in at least one body area. The most common area is the underarms (axilla), followed by the face (42%), palms of the hands (40%), and the soles of the feet (38%). It is estimated that nearly half (49%) of people with hyperhidrosis have not discussed their condition with a healthcare professional, either because they do not yet know it is a medical condition or believe that no adequate treatment options exist. Furthermore, in one survey, 75% of subjects with hyperhidrosis said that it has had negative impacts on their professional and social lives, sense of well-being, and emotional and mental health. We believe that, due to the lack of diagnosis and available treatment options, and general lack of knowledge about the disease, hyperhidrosis presents a substantial market opportunity for a new, innovative, effective, well-tolerated, topical treatment. We believe such a therapy could not only further penetrate the segment of patients who currently seek treatment from a physician, but also encourage more patients to seek treatment for this condition that causes them to deal with (and try to hide) it each and every day.
Current Hyperhidrosis Treatment Options and Limitations
The market for products to control sweating is large and highly underpenetrated by innovative prescription pharmaceutical products thoroughly tested in clinical trials. More specifically, current hyperhidrosis treatment options generally fall into one of the following categories:
• | Self-administered topicals, which include topical antiperspirants, some of which are prescription only, containing metal salts like aluminum that block the release of sweat to the skin surface by clogging the opening of the duct and Qbrexza® (glycopyrronium), approved in June 2018 by the FDA for the topical treatment of primary axillary hyperhidrosis in adult and pediatric patients nine years of age and older. For decades, topical antiperspirants containing metal salts have been the most widely used treatment option for hyperhidrosis. Over-the-counter (“OTC”) antiperspirants contain low concentrations of metal salts and are generally well-tolerated but limited in efficacy. Prescription antiperspirants containing higher concentrations of metal salts are typically recommended as the treatment of choice when OTC antiperspirants are ineffective. However, these are only marginally more effective, and their tolerability is |
33
limited by skin irritation associated with increased metal salt concentrations, which react with water to form irritating hydrochloric acid on the skin. Qbrexza is administered by prescription using a single-use cloth pre-moistened with the active ingredient, 2.4% glycopyrronium solution, packaged in individual pouches. Qbrexza inhibits the action of acetylcholine on sweat glands, thereby reducing sweating. While Qbrexza has shown to be effective in treating hyperhidrosis in certain subjects, we believe that there is room for improved and more sustained efficacy, as well as products that may potentially result in a lower incidence of unwanted systemic anticholinergic side effects including, but not limited to, dry mouth, blurred vision, and urinary hesitancy. We also believe that it may be attractive to patients to have other ways to deliver the drug through alternative formulations and/or delivery devices.
• | Injectable, systemic, and other treatments that block activation of the sweat glands. Therapeutic options for patients who are not satisfied with topical therapies are largely limited to more cumbersome or invasive treatment strategies directed either to blocking the activation of, destroying, or removing altogether the sweat glands. Intradermal injections of botulinum toxin, or BOTOX®, a neurotoxin that blocks the release of acetylcholine, are effective but can be painful, costly, and must be administered by a physician with patients receiving on average 20 to 40 injections to each arm pit every six to nine months. A microwave device, MiraDry®, is designed to overheat and destroy sweat glands as a different option. However, treatment with MiraDry® may be painful, require multiple physician visits, cause permanent destruction of the sweat gland and is not generally covered by insurance. All these treatments are time-consuming and require a significant investment of physician training and administration time and, in the case of microwave treatment, capital investment by the treating physician. As a result, these treatments have limited attractiveness both to doctors and their patients. Furthermore, they are also not approved or well-suited for application to the hands or feet. In contrast, we believe sofpironium bromide may potentially be developed for hands and feet. Iontophoresis, which involves soaking the hands or feet in water through which an electrical current is passed, can be performed in a physician’s office or at home, but requires repeated, time-consuming and often bothersome treatments. |
• | Surgical and other procedures intended to destroy or remove sweat glands. Some patients with severe hyperhidrosis may choose to be treated with invasive surgical techniques that involve removal of sweat glands or destruction of nerves that transmit activating signals to the glands. Surgery is a significant and costly permanent undertaking that can be associated with numerous severe side effects, including increased compensatory sweat production in other body areas. |
Deciding among these available treatments depends on many factors including the affected area, severity of the disease and impact on the patient’s quality of life due to the disease being uncontrolled. As a result of the limitations of these currently available treatment options, we believe that there is a significant unmet patient need for a new, effective, safe, well-tolerated, self-administered, prescription topical hyperhidrosis therapy.
Sofpironium Bromide for Hyperhidrosis
Sofpironium bromide is a potentially best-in-class topical anticholinergic product candidate we are developing for once-daily treatment of primary axillary hyperhidrosis in adult and pediatric patients nine years of age and older. Sofpironium bromide was designed as a structural analog of a well-known potent anticholinergic, glycopyrrolate, to achieve its therapeutic effect at the application site (skin) similar to glycopyrrolate. However, it differs from glycopyrrolate in that sofpironium bromide was retrometabolically designed. Retrometabolic drugs are intended to exert their action topically and are potentially rapidly metabolized once absorbed into the blood. This retrometabolic approach to drug design is intended to allow for highly effective doses to be used while limiting systemic side effects.
Key attributes of a retrometabolic drug include:
• | The synthesis of a retrometabolic drug is achieved by starting with a known inactive metabolite of a known active drug (e.g., glycopyrrolate). |
• | The inactive, or less active, metabolite is then structurally modified to an active form (an analogue of active drug in this case; glycopyrrolate) that will undergo a predictable one-step transformation back into the inactive metabolite in vivo. |
34
• | Thus, the retrometabolic drug concept is based upon predictable metabolic deactivation processes by enzymes found predominantly in the systemic circulation. |
Sofpironium bromide is delivered as a gel formulation in a metered-dose pump with an applicator that allows patients to avoid unwanted direct contact to the hands or other non-axillary body parts. We believe that this will help avoid certain side effects that could be caused by the unintended transference the drug such as to the eyes.
Clinical Development of Sofpironium Bromide
We, together with our partner Kaken, have conducted 19 clinical trials of sofpironium bromide gel that encompass over 1,200 subjects in the United States and Japan. These trials have evaluated the safety, tolerability, PK, and efficacy of sofpironium bromide gel in adult and pediatric primary hyperhidrosis patients and healthy adult subjects.
In clinical studies conducted to date, all three concentrations of sofpironium bromide gel tested (5%, 10%, and 15%) were safe and well tolerated. Treatment-emergent adverse events (“TEAEs”) were mostly mild or moderate in severity. There has been one death unrelated to sofpironium bromide and no serious adverse reactions have been reported in any clinical studies with sofpironium bromide gel. Twelve serious adverse events have been reported and all were determined to be unrelated to sofpironium bromide gel administration. Consistent with a retrometabolic drug design, a low incidence of systemic TEAEs has been found in all clinical studies of sofpironium bromide gel with a trend toward dose-dependency observed. The most common TEAEs were dry mouth and blurred vision. Of note, the TEAEs were predominantly mild or moderate in severity and transient in duration (i.e., resolving gradually with continued use). Local application site tolerability reactions of burning, itching, pain, erythema, and dryness at the axillae were predominantly minimal in severity and typically transient.
Overall, all three sofpironium bromide gel concentrations, 5%, 10%, and 15%, exhibited a larger absolute mean reduction in gravimetric sweat production (“GSP”) from baseline to end of treatment (“EOT”) compared with vehicle, with the reduction with the 15% concentration being statistically significant. However, while there was a slight trend toward dose response, all gel concentrations were essentially similar in patient-reported outcome measures based on the Hyperhidrosis Disease Severity Measure-Axillary (“HDSM-Ax”), modified Dermatology Life Quality Index (“DLQI”), and Hyperhidrosis Disease Severity Score (“HDSS”). The HDSM-Ax responses were seen as early as Day 8 and remained consistent throughout the applicable treatment period.
Phase 2b U.S. Clinical Trial (BBI-4000-CL-203)
The Phase 2b U.S. clinical trial was a multicenter, randomized, double blind, vehicle-controlled clinical trial to evaluate the safety and efficacy of topically-applied sofpironium bromide gel, 5%, 10%, and 15%, in patients with primary axillary hyperhidrosis. The trial enrolled a total of 227 patients across 23 clinical sites in the United States, with patients randomized to either sofpironium bromide gel, 5% (n=57), 10% (n=57), 15% (n=56), or vehicle gel (placebo; n=57) who applied the assigned product to the axillae (underarms) once daily, at bedtime, for 42 days. The objectives of this trial were to evaluate (1) the effect of sofpironium bromide gel, 5%, 10%, and 15% on hyperhidrosis disease severity as it relates to HDSM-Ax, GSP, HDSS, and modified DLQI; and (2) the safety and local tolerability of sofpironium bromide gel, 5%, 10%, and 15%.
Changes in HDSM-Ax measures indicated statistically significant differences from placebo (vehicle gel) in all sofpironium gel dose groups with all methods of analysis. Statistically significant differences in favor of active treatment groups were observed as early as Day 8 and were sustained over time. A significant higher proportion of active treatment subjects had at least a 2-point change from baseline to EOT in HDSM-Ax-11 items scale (5% gel: 47.4%, p=0.007; 10% gel: 49.1%, p=0.006; 15% gel: 50.0%, p=0.002; vehicle: 22.8%). Larger absolute mean reductions in GSP from baseline to EOT were found for all sofpironium bromide gel concentrations compared to vehicle gel, with the results with sofpironium bromide gel, 15% being statistically significant. Treatment with sofpironium bromide gel, 15% (pivotal Phase 3 active dose group) resulted in statistically significant reduction in GSP from baseline to EOT (-217 mg, p=0.06; vehicle -143 mg). The 5% and 10% dose groups resulted in -163 mg (p=0.32) and -174 mg (p=0.26) reduction in GSP from baseline to EOT, respectively. Consistently, superior ranked values indicating GSP reduction from baseline to EOT were observed for sofpironium bromide gel, 15% in comparison to vehicle. The ranked order analysis did not indicate a baseline to EOT reduction in GSP for the vehicle group; a p-value of 0.04 comparing sofpironium bromide, 15% gel to vehicle indicated the sofpironium bromide, 15% improvement to be real and not observed by chance. All sofpironium bromide gel groups met the secondary efficacy endpoints for HDSS and modified DLQI. It was prespecified in the study protocol and statistical analysis
35
plan that as a Phase 2 study, a 1-sided p<0.10 in favor of an active treatment would be regarded as statistically significant. All p-values cited in this study were 1-sided per the protocol and statistical analysis plan.
Among the safety population (includes all subjects who received study drug at least once; n=225), the subject incidence of TEAEs was higher in the sofpironium bromide gel, 15% group (51.9%) compared to the other groups (5% gel, 29.8%; 10% gel, 33.6%; vehicle gel, 15.8%). The majority of the systemic TEAEs were consistent with adverse events due to anticholinergic activity. The most common TEAEs included dry mouth (5% gel, 15.8%; 10% gel, 17.5%; 15% gel, 22.2% and vehicle gel, 1.8%) and blurred vision (5% gel, 3.5.%; 10% gel, 10.5%; 15% gel, 9.3% and vehicle gel, 0.0%). The majority of TEAEs in each group were mild or moderate in severity. Severe TEAEs were reported by 4 subjects in the 15% group and 2 subjects each in the 10% and 5% groups. The vast majority of severe TEAEs were anticholinergic TEAEs (dry mouth and vision blurred) or application site TEAEs (application site pain, application site pruritus, application site erythema, application site dryness, and application site exfoliation). There was one case of osteomyelitis which was severe and an SAE and was not related to sofpironium bromide. Treatment in all dose groups was well-tolerated. Local tolerability assessments indicated that all three active treatment groups; 15%, 10%, and 5% were well tolerated over the 42-day treatment period. Each local tolerability symptom/sign (burning, itching, dryness, scaling, and erythema) was absent in the majority of subjects in each group at each study visit. The incidence of these symptoms/signs was generally higher in the sofpironium bromide gel groups compared to the vehicle group. The majority of tolerability symptoms/signs were minimal to mild in severity and most resolved by the Day 57 visit. Severe tolerability symptoms/signs (burning, itching, and erythema) were reported only in the sofpironium gel groups.
Phase 3 Clinical Trials
Kaken has completed its pivotal Phase 3 clinical trial in subjects with primary axillary hyperhidrosis in Japan and achieved statistical significance (p<0.05) for primary and all secondary efficacy endpoints. In January 2020, we announced that Kaken submitted a new drug application for approval of manufacturing and marketing for sofpironium bromide in Japan for primary axillary hyperhidrosis.
Based on the positive results in the clinical trials conducted by us and Kaken to date, we intend to initiate two pivotal Phase 3 clinical trials in up to 350 subjects per trial with primary axillary hyperhidrosis in the United States, subject to the successful resolution of our dispute with the Bodor Plaintiffs and obtaining substantial additional funding. Assuming the results of the Phase 3 clinical trials are favorable, we plan thereafter to submit an NDA to the FDA for the treatment of hyperhidrosis.
BBI-3000 for the Potential Oral Treatment of Cutaneous T-Cell Lymphoma (CTCL)
BBI-3000 was designed as a potentially highly selective and safer (compared to Targretin®) RXR retinoid agonist currently under development for retinoid responsive skin conditions. While we believe there are several skin indications for which a novel RXR retinoid agonist, such as BBI-3000, may have therapeutic effect (e.g., psoriasis, photo-aging and CTCL), we are currently evaluating the development of BBI-3000 as a potentially better tolerated retinoid for the oral treatment of CTCL.
Retinoids are derivatives of vitamin A that play a pivotal role in a diverse group of biologic processes including, but not limited to, cellular proliferation, differentiation, apoptosis, and development. The biological activity and tolerability of the retinoid depends in part on the binding availability to RAR and RXR receptors. There are several topical retinoids and oral retinoids currently on the market that have shown efficacy in the treatment of several skin conditions, such as CTCL (e.g., bexarotene/Targretin®) and acne and psoriasis (e.g., tazarotene, adapalene and tretinoin). A common adverse reaction with the use of bexarotene is hyperlipidemia (abnormally elevated levels of lipids in the blood). Based on current data, we do not expect that BBI-3000 will cause significant hyperlipidemia. BBI-3000 may provide potential improvements to the treatment of CTCL by decreasing the incidence of systemic side effects while retaining the efficacy associated with systemic retinoid use. We believe that new oral retinoid treatments, such as BBI-3000, with potentially improved tolerability, and efficacy comparable to Targretin®, would be welcome in the marketplace.
Clinical Development of BBI-3000
BBI-3000 has been well tolerated in two Phase 1 studies (a single dose study and a multiple dose study) conducted by the NCI in healthy volunteers. There is an ongoing Phase 1b trial being conducted by the NCI to assess the biological effect of BBI-3000 on early stage breast cancer.
36
BBI-6000 for the Potential Topical Treatment of Psoriasis
BBI-6000 is a novel small molecule retinoic acid-related orphan nuclear receptor gamma (“RORg”) inhibitor we are developing as a potential prescription topical treatment for psoriasis. We believe that RORg inhibitors possess the potential to inhibit Th17 cell differentiation and reduce IL-17 production. BBI-6000 has been shown to exhibit specific effects on Th17-cell differentiation and has demonstrated selectivity for RORg. Additionally, studies with mouse Th17 cells have demonstrated that the compound suppresses IL-17A with no effect on interferon and in preclinical pharmacology screening, BBI-6000 specifically exhibited strong inhibition of IL-17A expression. Given the proposed role of BBI-6000 and RORg on IL-17 cytokine production, we believe BBI-6000 to have potential against a wide range of autoimmune diseases, such as psoriasis.
Preclinical Development
BBI-6000 is currently in the preclinical stages of development, with drug substance manufacturing, preclinical pharmacology testing and pre-formulation studies having been completed to date.
Competition
Our industry is highly competitive and subject to rapid and significant change. While we believe that our team’s extensive development and commercialization pharmaceutical experience in launching blockbuster drugs across multiple therapeutic areas, scientific knowledge, and global industry relationships provide us with competitive advantages, we face competition from pharmaceutical and biotechnology companies, including specialty pharmaceutical companies, as well as generic drug companies, over-the-counter companies, academic institutions, government agencies and research institutions.
Many of our competitors have significantly greater financial, technical and human resources than we do. Mergers and acquisitions in the pharmaceutical and biotechnology industries may result in even more resources being concentrated amongst a smaller number of our competitors. Our commercial opportunity could be reduced or eliminated if our competitors develop or market products or other novel therapies that are more effective, safer or less costly than our current or future product candidates or obtain regulatory approval for their products more rapidly than we may obtain approval for our product candidates. Our success will be based in part on our ability to identify, develop and manage a patented portfolio of product candidates that are safer and more effective than competing products and which will transform patient lives suffering from debilitating skin orders that are chronic and do not go away even with conventional treatment options.
Competition in Hyperhidrosis
If approved for the treatment of primary axillary hyperhidrosis, we anticipate that sofpironium bromide would compete with other therapies used for hyperhidrosis, including:
• | Self-Administered Treatments. Self-administered treatments, such as OTC and prescription topical antiperspirants, and Qbrexza® (glycopyrronium) 2.4% topical cloths. Oral and compounded topical anticholinergics could be used off-label by the administering physician. |
• | Non-Surgical Office-Based Procedures. Office-based procedures have been approved for the treatment of hyperhidrosis, including intradermal injections of BOTOX®, marketed by Allergan plc., and MiraDry®, a microwave-based treatment marketed by Miramar Labs, Inc. |
• | Surgical Treatments. Surgical treatments include techniques for the removal of sweat glands, such as excision, curettage and liposuction. Surgical procedures, such as endoscopic thoracic sympathectomy, are also used to destroy nerves that transmit activating signals to sweat glands. |
In addition to approved hyperhidrosis treatments, there are also several treatments under development that could potentially be used to treat hyperhidrosis and may compete with sofpironium bromide.
Intellectual Property and In-Licensing Agreements
Our success depends in large part upon our ability to secure proprietary protection for our products and technologies, including those in development, and to operate without infringing the proprietary rights of others. We seek to
37
avoid the latter by monitoring patents and publications that may affect our business, and to the extent we identify such threats, evaluate and take appropriate courses of action.
Patents extend for varying periods according to the date of patent filing or grant and the legal term of patents in various countries where patent protection is obtained. The actual protection afforded by a patent, which can vary from country to country, depends on the type of patent, the scope of its coverage and the availability of legal remedies in the country.
We also intend to use regulatory exclusivity (also called data package exclusivity) as a means of acquiring intellectual property protections that are separate and distinct to patents. This kind of right involves being given exclusivity for varying periods of time depending on the country to incentivize innovators who invest in and conduct clinical trials to produce data to demonstrate a drug is safe and effective for its intended use(s) and, as such, the data package in an NDA for the FDA should receive protection even if no patent is available. Other countries to varying extents do the same. In addition, there are other forms of intellectual property protection we may seek worldwide, including but not limited to trademarks, copyrights, trade secrets, orphan drug protection, pediatric exclusivity and the like, where available and appropriate for our business interests.
We further protect our proprietary information by requiring our directors, officers, employees, consultants, contractors and other advisors to execute nondisclosure and assignment of invention agreements upon commencement of their respective employment or engagement. Agreements with our employees also prevent them from bringing the proprietary rights of third parties to our company without adequate permission to do so. In addition, we require confidentiality or service agreements from third parties that receive our confidential information or materials.
As of December 31, 2019, regarding our complete patent portfolio, we own or possess an exclusive license to 23 issued U.S. patents and 52 issued foreign patents, which include granted European patent rights that have been validated in various EU member states. We also own or possess an exclusive license to eight pending U.S. patent applications and 89 pending international and foreign patent applications. With regard to our lead product candidate, sofpironium bromide, we own or possess an exclusive license to seven U.S. and 20 foreign patents as well as seven pending U.S. and 27 foreign patent applications which, if issued, may provide patent term coverage until 2040.
We also use other forms of protection besides regulatory exclusivity, such as trademark, copyright, and trade secret protection, to enhance our intellectual property, particularly where we do not believe patent protection is appropriate or obtainable. We aim to take advantage of all of the intellectual property rights that are available to us and believe that this comprehensive approach will provide us with proprietary exclusive positions for our product candidates, where available.
Manufacturing and Supply
We currently contract with third parties for the manufacture of its small-molecule drug substances and drug products for preclinical studies and clinical trials and intends to continue to do so in the future. To our knowledge, all of our clinical drug product manufacturing activities are in compliance with current good manufacturing practice (“cGMP”). We have assembled a team of experienced employees and consultants to provide the necessary technical, quality and regulatory oversight over the contract manufacturing organizations (“CMOs”) with which we contract. We rely on third-party cGMP manufacturers for scale-up and process development work and to produce sufficient quantities of development product candidates for use in clinical and preclinical trials.
Employees
As of December 31, 2019, we had 15 regular full-time employees, including eight in research and development. From time to time, we retain independent contractors. None of our employees is represented by a labor union or covered by a collective bargaining agreement. We have not experienced any work stoppages, and we consider our relations with our employees to be excellent.
Facilities
Our corporate headquarters are located in Boulder, Colorado, where we occupy facilities totaling approximately 3,038 square feet under lease agreements that expire in October 2021. We use our current facilities primarily for research and development and general and administrative personnel.
38
Legal Proceedings
On October 23, 2019, Bodor notified us of its termination of the License Agreement. Bodor alleges that we materially breached the License Agreement resulting in its termination. In connection with the termination, on October 23, 2019, the Bodor Plaintiffs filed the Bodor Complaint against us in the United States District Court for the Southern District of Florida. The Bodor Complaint alleges damages incurred by the Bodor Plaintiffs in connection with our alleged breach of the License Agreement. The Bodor Complaint seeks: (i) a declaratory judgment that the termination of the License Agreement by the Bodor Plaintiffs was valid and enforceable; (ii) an injunction requiring us to cease and desist use of the Bodor Plaintiffs’ intellectual property; and (iii) damages for breach of contract and breach of the covenant of good faith and fair dealing.
On October 30, 2019, we initiated an arbitration proceeding pursuant to Article 9 of the License Agreement with the AAA in Florida against the Bodor Plaintiffs. This arbitration seeks a declaratory judgment that the purported termination of the License Agreement by the Bodor Plaintiffs was invalid and unenforceable and asserts (i) a claim for breach of the License Agreement against the Bodor Plaintiffs, and (ii) a claim against the Bodor Plaintiffs for tortious interference with our business relations. We requested expedited treatment of the arbitration proceeding and concurrent mandatory mediation under the AAA rules. On October 30, 2019, we concurrently filed with the United States District Court for the Southern District of Florida a motion to dismiss (or stay) the complaint brought against us by the Bodor Plaintiffs described above. In its response to our motion to dismiss or, alternatively, to stay the litigation, Bodor agreed to comply with the License Agreement terms, which require mediation and, if necessary, arbitration to address disputes. Bodor also asked the court to stay the federal judicial proceedings against Brickell pending mediation, and, if necessary, final, conclusive and binding arbitration. We proceeded with mediation on December 10, 2019, which was unsuccessful. As a result, we recommenced the arbitration proceeding with the Bodor Plaintiffs to resolve the dispute. We continue to believe that the claims and assertions made by the Bodor Plaintiffs are in substance without merit.
The outcome of arbitration or litigation is inherently uncertain. If one or more of the legal claims made by Bodor were resolved against us, we may become subject to additional litigation claims, and our ability to continue to develop sofpironium bromide and our financial condition and operating results could be materially adversely affected. While we maintain insurance coverage for certain types of claims, such insurance coverage may be insufficient to cover all losses or all types of claims that may arise.
Although we do not believe the action is likely to be material, nor that the claims will be determined to be meritorious, Dr. Patricia S. Walker, our former President and Chief Scientific Officer, commenced litigation against us, an officer, our Board Chairperson and others, alleging wrongful termination for unspecified damages, claiming discrimination based on age, gender, and association with a person with a disability. We will contest these claims vigorously.
From time to time, we may become involved in other legal proceedings arising in the ordinary course of our business. We are not presently a party to any other legal proceedings that, if determined adversely to us, would individually or taken together have a material adverse effect on Brickell.
39
USE OF PROCEEDS
Unless otherwise indicated in any prospectus supplement, we intend to use the net proceeds from the sale of the securities under this prospectus for general corporate purposes, including clinical trial expenses, research and development expenses, general and administrative expenses, and potential partnerships with or acquisitions of companies and acquisitions or licensing of technologies that complement sofpironium bromide.
40
DESCRIPTION OF SECURITIES WE MAY OFFER
This prospectus contains a summary description of the common stock, preferred stock, debt securities, warrants and units that we may offer from time to time. As further described in this prospectus, these summary descriptions are not meant to be complete descriptions of each security. The particular terms of any security will be described in the accompanying prospectus supplement and other offering material. The accompanying prospectus supplement may update, change or add to the terms and conditions of the securities as described in this prospectus.
DESCRIPTION OF CAPITAL STOCK
The following description of our capital stock, together with the additional information we include in any applicable prospectus supplements, summarizes the material terms and provisions of the common stock and preferred stock that we may offer under this prospectus. The following description of our capital stock does not purport to be complete and are subject to, and qualified in their entirety by, our amended and restated certificate of incorporation and our amended and restated bylaws, and applicable law.
As of February 7, 2020, our restated certificate of incorporation authorizes us to issue 50,000,000 shares of common stock, par value $0.01 per share, and 5,000,000 shares of preferred stock, par value $0.01 per share. As of February 7, 2020, 8,702,261 shares of common stock were outstanding, along with (i) warrants issued and outstanding to purchase one share of our common stock at an exercise price of $0.07 per share, 490,683 shares of our common stock at an exercise price of $10.36 per share and 9,005 shares of our common stock at an exercise price of $33.31 per share; and (ii) we have 1,793,602 options issued and outstanding to purchase our common stock at a weighted average exercise price of $13.00 per share. No shares of preferred stock were outstanding.
Common Stock
The holders of our common stock are entitled to one vote for each share held of record on all matters submitted to a vote of our stockholders. The holders of our common stock are entitled to receive ratably the dividends, if any, that may be declared from time to time by our board of directors out of funds legally available for such dividends. In the event of a liquidation, dissolution or winding up of the Company, the holders of our common stock would be entitled to share ratably in all assets remaining after payment of liabilities and the satisfaction of any liquidation preferences granted to the holders of any outstanding shares of preferred stock.
Holders of our common stock have no preemptive rights and no conversion rights or other subscription rights. There are no redemption or sinking fund provisions applicable to our common stock. All the outstanding shares of common stock are, and all shares of common stock offered, when issued and paid for, will be validly issued, fully paid and nonassessable. The rights, preferences and privileges of holders of our common stock are subject to, and may be adversely affected by, the rights of the holders of any shares of our preferred stock.
Preferred Stock
Under our restated certificate of incorporation, our board of directors is authorized to issue shares of our preferred stock from time to time, in one or more classes or series, without stockholder approval. Prior to the issuance of shares of each class or series, our board of directors is required by the Delaware General Corporation Law (“DGCL”) and our restated certificate of incorporation to adopt resolutions and file a certificate of designation with the Delaware Secretary of State. The certificate of designation fixes for each class or series the designations, powers, preferences, rights, qualifications, limitations and restrictions of that class or series, including the following:
• | the number of shares constituting each class or series; |
• | voting rights; |
• | rights and terms of redemption, including sinking fund provisions; |
• | dividend rights and rates; |
• | terms concerning the distribution of assets; |
41
• | conversion or exchange terms; |
• | redemption prices; and |
• | liquidation preferences. |
All shares of preferred stock offered, when issued and paid for, will be validly issued, fully paid and nonassessable and will not have any preemptive or subscription rights.
We will specify the following terms relating to any class or series of preferred stock offered by us:
• | the title and stated value of the preferred stock; |
• | the number of shares of the preferred stock offered, the liquidation preference per share and the offering price of the preferred stock; |
• | the dividend rate(s), period(s) or payment date(s) or method(s) of calculation applicable to the preferred stock; |
• | whether dividends are cumulative or non-cumulative and, if cumulative, the date from which dividends on the preferred stock will accumulate; |
• | our right, if any, to defer payment of dividends and the maximum length of any such deferral period; |
• | the procedures for auction and remarketing, if any, for the preferred stock; |
• | the provisions for a sinking fund, if any, for the preferred stock; |
• | the provision for redemption, if applicable, of the preferred stock; |
• | any listing of the preferred stock on any securities exchange; |
• | the terms and conditions, if applicable, upon which the preferred stock will be convertible into common stock, including the conversion price or manner of calculation and conversion period; |
• | voting rights, if any, of the preferred stock; |
• | whether interests in the preferred stock will be represented by depositary shares; |
• | a discussion of any material or special United States federal income tax considerations applicable to the preferred stock; |
• | the relative ranking and preferences of the preferred stock as to dividend rights and rights upon the liquidation, dissolution or winding up of our affairs; |
• | any limitations on issuance of any class or series of preferred stock ranking senior to or on a parity with the class or series of preferred stock as to dividend rights and rights upon the liquidation, dissolution or winding up of our affairs; and |
• | any other specific terms, preferences, rights, limitations or restrictions of the preferred stock. |
Warrants
As of February 7, 2020, we had outstanding warrants to purchase (i) one share of our common stock at an exercise price of $0.07 per share, (ii) 490,683 shares of our common stock at an exercise price of $10.36 per share and (iii) 9,005 shares of our common stock at an exercise price of $33.31 per share.
42
Anti-Takeover Provisions
Delaware Anti-Takeover Law
We are subject to Section 203 of the DGCL. Section 203 generally prohibits a public Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder, unless:
• | prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction that resulted in the stockholder becoming an interested stockholder; |
• | the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the number of shares outstanding (a) shares owned by persons who are directors and also officers of the corporation and (b) shares issued under employee stock plans under which employee participants do not have the right to determine whether shares held subject to the plan will be tendered in a tender or exchange offer; or |
• | on or subsequent to the date of the transaction, the business combination is approved by the board and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock that is not owned by the interested stockholder. |
Section 203 defines a business combination to include:
• | any merger or consolidation involving the corporation and the interested stockholder; |
• | any sale, transfer, pledge or other disposition involving the interested stockholder of 10% or more of the assets of the corporation; |
• | subject to exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder; |
• | any transaction involving the corporation that has the effect of increasing the proportionate share of its stock owned by the interested stockholder; or |
• | the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation. |
In general, Section 203 of the DGCL defines an interested stockholder as any entity or person beneficially owning 15% or more of the outstanding voting stock of the corporation and any entity or person affiliated with or controlling or controlled by the entity or person.
Certificate of Incorporation and Bylaws
Some provisions of our restated certificate of incorporation and amended and restated bylaws could also have anti-takeover effects. These provisions:
• | provide for a board comprised of three classes of directors with each class serving a staggered three-year term; |
• | authorize our board of directors to issue preferred stock from time to time, in one or more classes or series, without stockholder approval; |
• | require the approval of at least two-thirds of our outstanding voting stock to amend specified provisions of our restated certificate of incorporation; |
• | require the approval of at least two-thirds of our total number of authorized directors, or two-thirds of our outstanding voting stock, to amend our amended and restated bylaws; |
43
• | provide that special meetings of our stockholders may be called only by our Chief Executive Officer, or by our board of directors pursuant to a resolution adopted by a majority of the total number of authorized directors; and |
• | do not include a provision for cumulative voting for directors (under cumulative voting, a minority stockholder holding a sufficient percentage of a class of shares may be able to ensure the election of one or more directors). |
The Nasdaq Capital Market Listing
Our common stock is listed on The Nasdaq Capital Market under the symbol “BBI.”
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is Computershare Trust Company, N.A. Its address is 250 Royall Street, Canton, Massachusetts 02021 and its telephone number is (800) 522-6645.
44
DESCRIPTION OF DEBT SECURITIES
We may issue debt securities under one or more trust indentures to be executed by us and a specified trustee. The terms of the debt securities will include those stated in the indenture and those made a part of the indenture by reference to the Trust Indenture Act of 1939 (the “Trust Indenture Act”). The indentures will be qualified under the Trust Indenture Act.
The following description sets forth certain anticipated general terms and provisions of the debt securities to which an accompanying prospectus supplement may relate. The particular terms of the debt securities offered by an accompanying prospectus supplement (which terms may be different than those stated below) and the extent, if any, to which such general provisions may apply to the debt securities so offered will be described in the prospectus supplement relating to such debt securities. Accordingly, for a description of the terms of a particular issue of debt securities, investors should review both the accompanying prospectus supplement relating thereto and the following description. A form of the indenture (as discussed herein) has been filed as an exhibit to the registration statement of which this prospectus is a part.
The debt securities will be our direct obligations and may be either senior debt securities or subordinated debt securities. The indebtedness represented by subordinated securities will be subordinated in right of payment to the prior payment in full of our senior debt (as defined in the applicable indenture).
Except as set forth in the applicable indenture and described in an accompanying prospectus supplement relating thereto, the debt securities may be issued without limit as to aggregate principal amount, in one or more series, secured or unsecured, in each case as established from time to time in or pursuant to authority granted by a resolution of the board of trustees or as established in the applicable indenture. All debt securities of one series need not be issued at the same time and, unless otherwise provided, a series may be reopened, without the consent of the holders of the debt securities of such series, for issuance of additional debt securities of such series.
The accompanying prospectus supplement relating to any series of debt securities being offered will contain their specific terms, including, without limitation:
• | their title and whether they are senior securities or subordinated securities; |
• | their initial aggregate principal amount and any limit on their aggregate principal amount; |
• | the percentage of the principal amount at which they will be issued and, if other than 100% of the principal amount, the portion of the principal amount payable upon declaration of acceleration of their maturity; |
• | the terms, if any, upon which they may be convertible or exchangeable into our common stock, other securities or other property and the terms and conditions upon which a conversion or exchange will be effected, including the initial conversion or exchange price or rate and the conversion or exchange period, any adjustments to the foregoing and any requirements relative to the reservation of shares for purposes of conversion or exchange; |
• | if convertible or exchangeable, any applicable limitations on the ownership or transferability of the common stock or preferred stock into which they are convertible or exchangeable; |
• | the date or dates, or the method for determining the date or dates, on which the principal will be payable; |
• | the rate or rates (which may be fixed or variable), or the method for determining the rate or rates, at which they will bear interest, if any; |
• | the date or dates, or the method for determining the date or dates, from which any interest will accrue, the interest payment dates on which any interest will be payable, the regular record dates for the interest payment dates, or the method by which the date will be determined and the basis upon which interest will be calculated if other than that of a 360-day year of twelve 30-day months; |
• | the place or places where the principal (and premium, if any) and interest, if any, will be payable, or the method of such payment, if by wire transfer, mail or other means; |
45
• | the period or periods within which, the price or prices at which and the terms and conditions upon which they may be redeemed, as a whole or in part, at our option, if we are to have the option; |
• | our obligation, if any, to redeem, repay or purchase them pursuant to any sinking fund or analogous provision or at the option of a holder, and the period or periods within which, the price or prices at which and the terms and conditions upon which they will be redeemed, repaid or purchased, as a whole or in part, pursuant to this obligation; |
• | if other than U.S. dollars, the currency or currencies in which they are denominated and in which any payments of principal (and premium, if any) or interest, if any, are payable, which may be a foreign currency or units of two or more foreign currencies or a composite currency or currencies, and the related terms and conditions; |
• | whether the payments of principal (and premium, if any) or interest, if any, may be determined with reference to an index, formula or other method (which index, formula or method may, but need not be, based on a currency, currencies, currency unit or units or composite currencies) and the manner in which the amounts will be determined; |
• | any additions to, modifications of or deletions from their terms with respect to the events of default, to the rights of the trustee or the holders to declare the principal amount thereof due and payable, or to the covenants, in each case as set forth in the indenture; |
• | any provisions for collateral security for their repayment; |
• | any provisions relating to guarantees; |
• | any trustees, depositories, interest rate calculation agents, exchange rate calculation agents or other agents; |
• | whether they will be issued in certificated or book-entry form; |
• | the date any temporary global security will be dated if other than the date of original issuance of the first security of such series to be issued; |
• | if issued in definitive form only upon receipt of certain certificates or other documents or satisfaction of other conditions, the form and/or terms of such certificates, documents or conditions; |
• | if to be issued upon the exercise of debt warrants, the time, manner and place to be authenticated and delivered; |
• | the denominations if other than $1,000 and any integral multiple thereof; |
• | the applicability, if any, of defeasance and covenant defeasance provisions of the applicable indenture; |
• | whether and under what circumstances we will pay additional amounts as contemplated in the applicable indenture in respect of any tax, assessment or governmental charge and, if so, whether we will have the option to redeem them in lieu of making the payment; and |
• | any other terms and any deletions from or modifications or additions to the applicable indenture. |
The debt securities may provide for less than the entire principal amount thereof to be payable upon declaration of acceleration of the maturity thereof. Special federal income tax, accounting and other considerations applicable to debt securities will be described in the accompanying prospectus supplement.
The applicable indenture may contain provisions that would limit our ability to incur indebtedness or that would afford holders of debt securities protection in the event of a highly leveraged or similar transaction involving us or in the event of a change of control.
46
Investors should review the accompanying prospectus supplement for information with respect to any deletions from, modifications of or additions to the events of default or covenants that are described below, including any addition of a covenant or other provision providing event risk or similar protection.
Merger, Consolidation or Sale
The applicable indenture will provide that we may consolidate with or merge into, or convey, transfer or lease all or substantially all of our assets to any other person (as defined therein), provided that:
• | we are the continuing entity, or the successor entity (if other than the Company) formed by or resulting from any consolidation or merger or which has received the transfer of our assets will be organized and validly existing under the laws of any U.S. domestic jurisdiction and expressly assumes our obligations on the applicable debt securities and under the indenture; |
• | immediately after giving effect to the transaction, no event of default under the applicable indenture, and no event which, after notice or the lapse of time, or both, would become an event of default, will have occurred and be continuing; and |
• | an officer’s certificate and legal opinion covering these conditions will be delivered to the trustee. |
Covenants
The applicable indenture will contain covenants requiring us to take certain actions and prohibiting us from taking certain actions. The covenants with respect to any series of debt securities will be described in the accompanying prospectus supplement.
Events of Default, Notice and Waiver
Each indenture will describe specific “events of default” with respect to a series of debt securities issued under the indenture. These “events of default” are likely to include (with grace and cure periods):
• | our failure to pay any installment of interest; |
• | our failure to pay the principal (or premium, if any) at maturity; |
• | our failure to make any required sinking fund payment; |
• | our breach of any other covenant or warranty contained in the applicable indenture (other than a covenant added to the indenture solely for the benefit of a different series of debt securities); and |
• | certain events of bankruptcy, insolvency or reorganization, or court appointment of a receiver, liquidator or trustee of us or any substantial part of our property. |
If an event of default resulting from certain events of bankruptcy described in the indenture occurs, all outstanding debt securities of that series will become due and payable immediately. If any other event of default under any indenture with respect to debt securities of any series at the time outstanding occurs and is continuing, then the applicable trustee or the holders of not less than 25% of the principal amount of the outstanding debt securities of that series may declare the principal amount (or, if the debt securities of that series are original issue discount securities or indexed securities, such portion of the principal amount as may be specified in the terms thereof) of all the debt securities of that series to be due and payable immediately by written notice thereof to us (and to the applicable trustee if given by the holders). However, at any time after such a declaration of acceleration with respect to debt securities of such series (or of all debt securities then outstanding under any indenture, as the case may be) has been made, the holders of not less than a majority in principal amount of outstanding debt securities of such series (or of all debt securities then outstanding under the applicable indenture, as the case may be) may rescind and annul such declaration and its consequences if:
• | the rescission would not conflict with any judgment or decree; and |
47
• | all events of default, other than the non-payment of accelerated principal, interest or premium (or specified portion thereof), with respect to debt securities of such series (or of all debt securities then outstanding under the applicable indenture, as the case may be) have been cured or waived as provided in such indenture. |
Each indenture also will provide that the holders of not less than a majority in principal amount of the outstanding debt securities of any series (or of all debt securities then outstanding under the applicable indenture, as the case may be) may waive any past default with respect to the series and its consequences, except a:
• | continuing payment default; or |
• | covenant default that cannot be modified or amended without the consent of the holder of each outstanding debt security affected thereby. |
Each trustee will be required to give notice to the holders of debt securities within a certain number of days of a default under the applicable indenture unless the default has been cured or waived; provided, however, that the trustee may withhold notice to the holders of any series of debt securities of any default with respect to the series (except a default in the payment of the principal of (or premium, if any) or interest on any debt security of the series or in the payment of any sinking fund installment in respect of any debt security of the series) if specified responsible officers of the trustee consider withholding the notice to be in the interest of the holders.
Each indenture will prohibit the holders of debt securities of any series from instituting any proceedings, judicial or otherwise, with respect to the indenture or for any remedy thereunder, except in the case of failure of the applicable trustee, for a certain period of time after the trustee has received a written request to institute proceedings in respect of an event of default from the holders of not less than a majority in principal amount of the outstanding debt securities of such series, as well as the furnishing of indemnity reasonably satisfactory to it.
This provision will not prevent any holder of debt securities from instituting a suit to enforce the payment of the principal of (and premium, if any) and interest on the debt securities at the respective due dates thereof.
Subject to the indenture, no trustee will be under any obligation to exercise any of its rights or powers under an indenture at the request or direction of any holders of any series of debt securities then outstanding, unless the holders furnish the trustee thereunder reasonable security or indemnity. The holders of not less than a majority in principal amount of the outstanding debt securities of any series (or of all debt securities then outstanding under an indenture, as the case may be) will have the right to direct the time, method and place of conducting any proceeding for any remedy available to the applicable trustee, or of exercising any trust or power conferred upon the trustee. However, a trustee may refuse to follow any direction, which is in conflict with any law or the applicable indenture, which may involve the trustee in personal liability or which may be unduly prejudicial to the holders of debt securities of such series not joining therein.
Within a certain period of time of the close of each fiscal year, we will be required to deliver to each trustee, a certificate, signed by one of several specified officers, stating whether or not the officer has knowledge of any default under the applicable indenture and, if so, specifying each default and the nature and status thereof.
Modification of the Indenture
The indenture will likely provide that it may be modified or amended, with the consent of the holders of not less than a majority in principal amount of each series of the outstanding debt securities issued under the indenture affected by the modification or amendment, provided that no modification or amendment may, without the consent of each affected holder of the debt securities:
• | change the stated maturity date or reduce the amount of the principal of (or premium, if any) or reduce the rate of interest or change the time for payment of any installment of interest, if any, on the debt securities; |
• | change the currency of payment of principal of (or premium, if any) or interest, if any, on the debt securities; |
48
• | waive a default or event of default in the payment of principal of (or premium, if any) or interest on the debt securities (other than as described in the indenture); |
• | waive a redemption payment, if any, or alter or waive any of the provisions in the indenture with respect to redemption; |
• | reduce the above-stated percentage of holders of the debt securities necessary to modify or amend the indenture; or |
• | modify the foregoing requirements or reduce the percentage of the outstanding debt securities necessary to waive compliance with certain provisions of the indenture or for waiver of certain defaults. |
A record date may be set for any act of the holders with respect to consenting to any amendment.
The holders of not less than a majority in principal amount of the outstanding debt securities of each series affected thereby will have the right to waive our compliance with certain covenants in the indenture. Each indenture will contain provisions for convening meetings of the holders of debt securities of a series to take permitted action. Under certain circumstances, we and the trustee may make modifications and amendments to an indenture without the consent of any holders of outstanding debt securities.
Conversion of Debt Securities
The terms and conditions, if any, upon which any debt securities are convertible or into our common stock or preferred stock will be set forth in the applicable accompanying prospectus supplement. The terms will include:
• | whether the debt securities are convertible into our common stock or preferred stock; |
• | the conversion price (or the manner of calculating the price); |
• | the conversion period; |
• | the events requiring an adjustment to the conversion price and provisions affecting conversion if the debt securities are redeemed; and |
• | any restrictions on conversion. |
Subordination
Upon any distribution to our creditors in a liquidation, dissolution or reorganization, the payment of the principal of and interest on any subordinated securities will be subordinated to the extent provided in the applicable indenture to the prior payment in full of all senior securities. No payment of principal or interest will be permitted to be made on subordinated securities at any time if any payment default or any other default which permits accelerations exists. After all senior securities are paid in full and until the subordinated securities are paid in full, holders of subordinated securities will be subrogated to the right of holders of senior securities to the extent that distributions otherwise payable to holders of subordinated securities have been applied to the payment of senior securities. By reason of any subordination, in the event of a distribution of assets upon our insolvency, some of our general creditors may recover more, ratably, than holders of subordinated securities. The accompanying prospectus supplement or the information incorporated herein by reference will contain the approximate amount of senior securities outstanding as of the end of our most recent fiscal quarter.
Global Debt Securities
The debt securities of a series may be issued in whole or in part in global form. The global securities will be deposited with a depositary, or with a nominee for a depositary, identified in the accompanying prospectus supplement. In this case, one or more global securities will be issued in a denomination or aggregate denominations equal to the portion of the aggregate principal amount of outstanding debt securities of the series to be represented by the global security or securities. Unless and until it is exchanged in whole or in part for debt securities in definitive form, a global security may not be transferred except as a whole by the depositary for the global security to a nominee of the depositary or by a nominee of
49
the depositary to the depositary or another nominee of the depositary or by the depositary or any nominee to a successor of the depositary or a nominee of the successor.
The specific material terms of the depositary arrangement with respect to any portion of a series of debt securities to be represented by a global security will be described in the applicable accompanying prospectus supplement. We anticipate that the following provisions will apply to all depositary arrangements.
Upon the issuance of a global security, the depositary for the global security will credit, on its book-entry registration and transfer system, the respective principal amounts of the debt securities represented by the global security to the accounts of persons or participants that have accounts with the depositary. The accounts to be credited will be designated by any underwriters or agents participating in the distribution of the debt securities. Ownership of beneficial interests in a global security will be limited to participants or persons that may hold interests through participants. Ownership of beneficial interests in the global security will be shown on, and the transfer of that ownership will be effected only through, records maintained by the depositary for the global security, with respect to interests of participants, or by participants or persons that hold through participants, with respect to interests of persons other than participants. So long as the depositary for a global security, or its nominee, is the registered owner of the global security, the depositary or the nominee, as the case may be, will be considered the sole owner or holder of the debt securities represented by the global security for all purposes under the indenture; provided, however, that for purposes of obtaining any consents or directions required to be given by the holders of the debt securities, we, the trustee and our agents will treat a person as the holder of the principal amount of debt securities as specified in a written statement of the depositary. Except as set forth herein or otherwise provided in the accompanying prospectus supplement, owners of beneficial interests in a global security will not be entitled to have the debt securities represented by the global security registered in their names, will not receive physical delivery of the debt securities in definitive form and will not be considered the owners or holders thereof under the indenture.
Principal, premium, if any, and interest payments on debt securities represented by a global security registered in the name of a depositary or its nominee will be made to the depositary or its nominee, as the case may be, as the registered owner of the global security. Neither we, the trustee nor any paying agent for the debt securities will have any responsibility or liability for any aspect of the records relating to or payments made on account of beneficial ownership interests in the global security or for maintaining, supervising or reviewing any records relating to the beneficial ownership interests.
We expect that the depositary for any debt securities represented by a global security, upon receipt of any payment of principal, premium, if any, or interest will immediately credit participants’ accounts with payments in amounts proportionate to their respective beneficial interests in the principal amount of the global security as shown on the records of the depositary. We also expect that payments by participants will be governed by standing instructions and customary practices, as is now the case with the securities held for the accounts of customers registered in “street names” and will be the responsibility of the participants.
If the depositary for any debt securities represented by a global security is at any time unwilling or unable to continue as depositary and a successor depositary is not appointed by us within the period of time set forth in the indenture, we will issue the debt securities in definitive form in exchange for the global security. In addition, we may at any time, and in our sole discretion, determine not to have any of the debt securities of a series represented by one or more global securities and, in that event, will issue debt securities of the series in definitive form in exchange for all of the global security or securities representing the debt securities.
The laws of some states require that certain purchasers of securities take physical delivery of the securities in definitive form. These laws may impair the ability to transfer beneficial interests in debt securities represented by global securities.
Governing Law
The indenture and the debt securities will be governed by and construed in accordance with the internal laws of the State of New York.
50
DESCRIPTION OF WARRANTS
We may issue warrants for the purchase of common stock, preferred stock or debt securities, and may issue warrants independently or together with common stock, preferred stock or debt securities, or attached to, or separate from, such securities. We will issue each series of warrants under a separate warrant agreement between us and a bank or trust company as warrant agent, as specified in the applicable prospectus supplement. The form of the warrant agreement and the form of the warrant certificate will be filed with the SEC and incorporated by reference as an exhibit to the registration statement of which this prospectus is a part.
The warrant agent will act solely as our agent in connection with the warrants and will not act for or on behalf of warrant holders. The following sets forth certain general terms and provisions of the warrants that may be offered under this registration statement. Further terms of the warrants and the applicable warrant agreement will be set forth in the applicable prospectus supplement.
The applicable prospectus supplement will describe the terms of the warrants in respect of which this prospectus is being delivered, including, where applicable, the following:
• | the title of such warrants; |
• | the aggregate number of such warrants; |
• | the price or prices at which such warrants will be issued; |
• | the type and number of securities purchasable upon exercise of such warrants; |
• | the designation and terms of the other securities, if any, with which such warrants are issued and the number of such warrants issued with each such offered security; |
• | the date, if any, on and after which such warrants and the related securities will be separately transferable; |
• | the price at which each security purchasable upon exercise of such warrants may be purchased; |
• | the date on which the right to exercise such warrants shall commence and the date on which such right shall expire; |
• | the minimum or maximum amount of such warrants which may be exercised at any one time; |
• | information with respect to book-entry procedures, if any; |
• | any anti-dilution protection; |
• | a discussion of certain U.S. federal income tax considerations; and |
• | any other terms of such warrants, including terms, procedures and limitations relating to the transferability, exercise and exchange of such warrants. |
Warrant certificates will be exchangeable for new warrant certificates of different denominations and warrants may be exercised at the corporate trust office of the warrant agent or any other office indicated in the applicable prospectus supplement. Prior to the exercise of their warrants, holders of warrants will not have any of the rights of holders of the securities purchasable upon such exercise or to any dividend payments or voting rights as to which holders of the shares of common stock or preferred stock purchasable upon such exercise may be entitled.
Each warrant will entitle the holder to purchase for cash such number of shares of common stock, preferred stock or debt securities, at such exercise price as shall, in each case, be set forth in, or be determinable as set forth in, the applicable prospectus supplement relating to the warrants offered thereby. After the expiration date set forth in the applicable prospectus supplement, unexercised warrants will be void.
51
Warrants may be exercised as set forth in the applicable prospectus supplement relating to the warrants.
Upon receipt of payment and the warrant certificate properly completed and duly executed at the corporate trust office of the warrant agent or any other office indicated in the applicable prospectus supplement, we will, as soon as practicable, forward the securities purchasable upon such exercise. If less than all of the warrants are presented for exercise with respect to a warrant certificate, a new warrant certificate will be issued for the remaining amount of warrants.
52
DESCRIPTION OF UNITS
We may issue units consisting of one or more of the other securities that may be offered under this prospectus, in any combination. Units may also include debt obligations of a third party. These units may be issuable as, and for a specified period of time may be transferable only as, a single security, rather than as the separate constituent securities comprising such units. The statements made in this section relating to the units are summaries only and are not complete. When we issue units, we will provide the specific terms of the units in a prospectus supplement. To the extent the information contained in the prospectus supplement differs from this summary description, you should rely on the information in the prospectus supplement.
When we issue units, we will provide in a prospectus supplement the following terms of the units being issued when applicable:
• | the title of any series of units; |
• | identification and description of the separate constituent securities comprising the units; |
• | the price or prices at which the units will be issued; |
• | the date, if any, on and after which the constituent securities comprising the units will be separately transferable; |
• | information with respect to any book-entry procedures; |
• | a discussion of any material or special U.S. federal income tax consequences applicable to an investment in the units; and |
• | any other material terms of the units and their constituent securities. |
53
PLAN OF DISTRIBUTION
We may sell the securities from time to time pursuant to underwritten public offerings, negotiated transactions, at the market offerings, block trades or a combination of these methods. We may sell the securities to or through underwriters or dealers, through agents, or directly to one or more purchasers.
We may distribute securities from time to time in one or more transactions:
• | at a fixed price, or prices, which may be changed from time to time; |
• | at market prices prevailing at the time of sale; |
• | at prices related to such prevailing market prices; or |
• | at negotiated prices. |
Unless stated otherwise in the applicable prospectus supplement, the obligations of any underwriter to purchase securities will be subject to certain conditions, and the underwriter will be obligated to purchase all of the applicable securities if any are purchased. If a dealer is used in a sale, we may sell the securities to the dealer as principal. The dealer may then resell the securities to the public at varying prices to be determined by the dealer at the time of resale.
We or our agents may solicit offers to purchase securities from time to time. Unless stated otherwise in the applicable prospectus supplement, any agent will be acting on a best efforts basis for the period of its appointment.
In connection with the sale of securities, underwriters or agents may receive compensation (in the form of discounts, concessions or commissions) from us or from purchasers of securities for whom they may act as agents. Underwriters may sell securities to or through dealers, and such dealers may receive compensation in the form of discounts, concessions or commissions from the underwriters and/or commissions from the purchasers for whom they may act as agents. Underwriters, dealers and agents that participate in the distribution of securities may be deemed to be underwriters, as that term is defined in the Securities Act, and any discounts or commissions received by them from us and any profits on the resale of the securities by them may be deemed to be underwriting discounts and commissions under the Securities Act. We will identify any such underwriter or agent, and we will describe any compensation paid to them, in the related prospectus supplement.
Underwriters, dealers and agents may be entitled under agreements with us to indemnification against and contribution toward certain civil liabilities, including liabilities under the Securities Act.
If stated in the applicable prospectus supplement, we will authorize agents and underwriters to solicit offers by certain specified institutions or other persons to purchase securities at the public offering price set forth in the prospectus supplement under delayed delivery contracts providing for payment and delivery on a specified date in the future. Institutions with whom these contracts may be made include commercial and savings banks, insurance companies, pension funds, investment companies, educational and charitable institutions, and other institutions, but shall in all cases be subject to our approval. These contracts will be subject only to those conditions set forth in the applicable prospectus supplement and the applicable prospectus supplement will set forth the commission payable for solicitation of these contracts. The obligations of any purchaser under any such contract will be subject to the condition that the purchase of the securities shall not be prohibited at the time of delivery under the laws of the jurisdiction to which the purchaser is subject. The underwriters and other agents will not have any responsibility in respect of the validity or performance of these contracts.
The securities may or may not be listed on a national securities exchange or traded in the over-the-counter market, as set forth in the applicable prospectus supplement. No assurance can be given as to the liquidity of the trading market for any of our securities. Any underwriter may make a market in these securities. However, no underwriter will be obligated to do so, and any underwriter may discontinue any market making at any time, without prior notice.
If underwriters or dealers are used in the sale, until the distribution of the securities is completed, SEC rules may limit the ability of any underwriters and selling group members to bid for and purchase the securities. As an exception to these rules, representatives of any underwriters are permitted to engage in certain transactions that stabilize the price of the securities. These transactions may consist of bids or purchases for the purpose of pegging, fixing or maintaining the price of the securities. If the underwriters create a short position in the applicable securities in connection with any offering (in other
54
words, if they sell more securities than are set forth on the cover page of the applicable prospectus supplement) the representatives of the underwriters may reduce that short position by purchasing securities in the open market. The representatives of the underwriters may also elect to reduce any short position by exercising all or part of any over-allotment option we may grant to the underwriters, as described in the prospectus supplement. The representatives of the underwriters may also impose a penalty bid on certain underwriters and selling group members. This means that if the representatives purchase securities in the open market to reduce the underwriters’ short position or to stabilize the price of the securities, they may reclaim the amount of the selling concession from the underwriters and selling group members who sold those shares as part of the offering.
In general, purchases of a security for the purpose of stabilization or to reduce a short position could cause the price of the security to be higher than it might be in the absence of those purchases. The imposition of a penalty bid might also have an effect on the price of the securities to the extent that it discourages resales of the securities. The transactions described above may have the effect of causing the price of the securities to be higher than it would otherwise be. If commenced, the representatives of the underwriters may discontinue any of the transactions at any time. In addition, the representatives of any underwriters may determine not to engage in those transactions or that those transactions, once commenced, may be discontinued without notice.
Certain of the underwriters or agents and their associates may engage in transactions with and perform services for us or our affiliates in the ordinary course of their respective businesses.
In no event will the commission or discount received by any Financial Industry Regulatory Authority (“FINRA”), member or independent broker-dealer participating in a distribution of securities exceed 8% of the aggregate principal amount of the offering of securities in which that FINRA member or independent broker-dealer participates.
55
INFORMATION INCORPORATED BY REFERENCE
The SEC allows us to incorporate by reference information into this document. This means that we can disclose important information to you by referring you to another document filed separately with the SEC. The information incorporated by reference is an important part of this prospectus, and information that we file later with the SEC will automatically update and supersede this information. We incorporate by reference the documents listed below and any future filings made with the SEC under Sections 13(a), 13(c), 14, or 15(d) of the Exchange Act made subsequent to the date of this prospectus until the termination of the offering of the securities described in this prospectus (other than information in such filings that was “furnished,” under applicable SEC rules, rather than “filed”).
We incorporate by reference the following documents or information that we have filed with the SEC:
• | our quarterly report on Form 10-Q for the quarter ended September 30, 2019 filed with the SEC on November 14, 2019; |
• | our current reports on Form 8‑K filed with the SEC on June 3, 2019, July 18, 2019, August 21, 2019, August 30, 2019, September 3, 2019, October 10, 2019, October 24, 2019, October 30, 2019, November 25, 2019, December 23, 2019 and February 10, 2020 (in each case, except for information contained therein which is furnished rather than filed); |
• | our definitive proxy statement on Schedule 14A (the “Proxy Statement”) filed with the SEC on July 12, 2019 (as amended by Amendment No. 1 to the Proxy Statement filed on August 8, 2019, Amendment No. 2 to the Proxy Statement filed on August 20, 2018 and Amendment No. 3 to the Proxy Statement filed on August 23, 2019); and |
• | the description of our common stock contained in our registration statement on Form S-8 filed with the SEC on September 10, 2019, including any amendment or report filed for the purpose of updating such description. |
Any statement contained in this prospectus or contained in a document incorporated or deemed to be incorporated by reference into this prospectus will be deemed to be modified or superseded to the extent that a statement contained in this prospectus or any subsequently filed supplement to this prospectus, or document deemed to be incorporated by reference into this prospectus, modifies or supersedes such statement
You may request a copy of these filings at no cost, by writing or telephoning us at the following address:
Brickell Biotech, Inc.
5777 Central Avenue
Suite 102
Boulder, CO 80301
(720) 505-4755
E-mail: investorrelations@brickellbio.com
You should rely only on the information incorporated by reference or provided in this prospectus or in any prospectus supplement. We have not authorized anyone else to provide you with different or additional information. An offer of these securities is not being made in any jurisdiction where the offer or sale is not permitted. You should not assume that the information in this prospectus or any prospectus supplement is accurate as of any date other than the date on the front of those documents.
WHERE YOU CAN FIND MORE INFORMATION
This prospectus is part of a registration statement we filed with the SEC. This prospectus does not contain all of the information set forth in the registration statement and the exhibits to the registration statement. For further information with respect to us and the securities we are offering under this prospectus, we refer you to the registration statement and the exhibits and schedules filed as a part of the registration statement. You should rely only on the information contained in this prospectus or incorporated by reference in this prospectus. We have not authorized anyone else to provide you with different information. We are not making an offer of these securities in any state where the offer is not permitted. You should not
56
assume that the information in this prospectus is accurate as of any date other than the date on the front page of this prospectus, regardless of the time of delivery of this prospectus or any sale of the securities offered by this prospectus.
We file annual, quarterly and current reports, proxy statements and other information with the SEC. Our SEC filings are available to the public from commercial document retrieval services and over the Internet at the SEC’s website at http://www.sec.gov.
Copies of certain information filed by us with the SEC are also available on our website at www.brickellbio.com. Information contained in or accessible through our website does not constitute a part of this prospectus and is not incorporated by reference in this prospectus.
57
LEGAL MATTERS
The validity of the common stock and certain other legal matters will be passed upon for us by Mayer Brown LLP, New York, New York.
EXPERTS
On August 31, 2019, the Delaware corporation formerly known as “Vical Incorporated”, completed a merger transaction in accordance with the terms and conditions of the Agreement and Plan of Merger and Reorganization, dated as of June 2, 2019, as amended by Amendment No. 1 to Agreement and Plan of Merger and Reorganization, dated August 20, 2019, and as further amended on August 30, 2019 (the “Merger Agreement”), by and among Vical Incorporated (“Vical”), Brickell and Victory Subsidiary, Inc., a wholly-owned subsidiary of Vical formed in connection with the merger (the “Merger Sub”), pursuant to which the Merger Sub merged with and into Brickell, with Brickell surviving the merger as a wholly-owned subsidiary of Vical (the “Merger”). On August 31, 2019, in connection with, and prior to, the consummation of the Merger, Vical effected a reverse stock split of its common stock, par value $0.01 per share, at a ratio of 1-for-7 (the “Reverse Stock Split”). Additionally, on August 31, 2019, immediately after the completion of the Merger, the Company changed its name from “Vical Incorporated” to “Brickell Biotech, Inc.”
The consolidated financial statements of Brickell Biotech, Inc. as of December 31, 2018 and 2017, and for each of the years in the two-year period ended December 31, 2018, appearing in Brickell Biotech, Inc.’s Current Report on Form 8-K, dated February 10, 2020, have been audited by Ernst & Young LLP, independent registered public accounting firm, as set forth in their report thereon (which contains an explanatory paragraph describing conditions that raise substantial doubt about the Company’s ability to continue as a going concern as described in Note 1 to the consolidated financial statements) included therein, and incorporated herein by reference. Such financial statements are, and audited financial statements to be included in subsequently filed documents will be, incorporated herein in reliance upon the reports of Ernst & Young LLP pertaining to such financial statements (to the extent covered by consents filed with the SEC) given on the authority of such firm as experts in accounting and auditing.
58
Common Stock
Preferred Stock
Debt Securities
Warrants
Units
Preferred Stock
Debt Securities
Warrants
Units
PROSPECTUS
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 14. Other Expenses of Issuance and Distribution.
The following table sets forth the costs and expenses, other than underwriting discounts and commissions, payable by us in connection with the sale and distribution of the securities being registered. All of the amounts shown are estimates:
Amount to be paid | |
SEC registration fee | $9,735 |
FINRA filing fees | 11,750 |
Printing expenses | * |
Legal fees and expenses | * |
Accounting fees and expenses | * |
Miscellaneous | * |
Total | $* |
* | These fees are calculated based on the number of issuances and the amount of securities offered and accordingly cannot be estimated at this time. |
Item 15. Indemnification of Directors and Officers.
The Company is incorporated under the laws of the State of Delaware. Section 145 of the Delaware General Corporation Law (the “DGCL”) provides that a Delaware corporation may indemnify any persons who are, or are threatened to be made, parties to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of such corporation), by reason of the fact that such person was an officer, director, employee or agent of such corporation, or is or was serving at the request of such person as an officer, director, employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by such person in connection with such action, suit or proceeding, provided that such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation’s best interests and, with respect to any criminal action or proceeding, had no reasonable cause to believe that his or her conduct was illegal. A Delaware corporation may indemnify any persons who are, or are threatened to be made, a party to any threatened, pending or completed action or suit by or in the right of the corporation by reason of the fact that such person was a director, officer, employee or agent of such corporation, or is or was serving at the request of such corporation as a director, officer, employee or agent of another corporation or enterprise. The indemnity may include expenses (including attorneys’ fees) actually and reasonably incurred by such person in connection with the defense or settlement of such action or suit provided such person acted in good faith and in a manner he or she reasonably believed to be in or not opposed to the corporation’s best interests except that no indemnification is permitted without judicial approval if the officer or director is adjudged to be liable to the corporation. Where an officer or director is successful on the merits or otherwise in the defense of any action referred to above, the corporation must indemnify him or her against the expenses which such officer or director has actually and reasonably incurred. Article XI of the Company’s amended and restated certificate of incorporation provides for indemnification of its directors and officers, and Article V of the Company’s amended and restated bylaws provides for indemnification of its directors, officers, employees and other agents, to the maximum extent permitted by the DGCL. In addition, the Company maintains a policy providing directors’ and officers’ liability insurance.
Section 102 of the DGCL permits a corporation to provide in its certificate of incorporation that a director of the corporation shall not be personally liable to the corporation or its stockholders for monetary damages for breach of fiduciary duties as a director, except for liability:
• | for any breach of the director’s duty of loyalty to the corporation or its stockholders; |
• | for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law; |
II-1
• | for acts related to unlawful stock repurchases, redemptions or other distributions or payment of dividends; or |
• | for any transaction from which the director derived an improper personal benefit. |
The Company’s amended and restated certificate of incorporation includes such a provision. Expenses incurred by any officer or director in defending any such action, suit or proceeding in advance of its final disposition shall be paid by the Company upon delivery of an undertaking, by or on behalf of such director or officer, to repay all amounts so advanced if it shall ultimately be determined that such director or officer is not entitled to be indemnified by the Company.
Item 16. Exhibits.
The list of exhibits following the signature page of this registration statement is incorporated by reference herein.
Item 17. Undertakings.
(1) | The undersigned registrant hereby undertakes: |
a. | To file, during any period in which offers or sales are being made, a post‑effective amendment to this registration statement: |
i. | To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933, as amended (the “Securities Act”); |
ii. | To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post‑effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the SEC pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement; and |
iii. | To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement; |
provided, however, that the undertaking set forth in paragraphs (1)(a)(i), (1)(a)(ii) and (1)(a)(iii) above do not apply if the information required to be included in a post‑effective amendment by those paragraphs is contained in reports filed with or furnished to the SEC by the registrant pursuant to Section 13 or Section 15(d) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”), that are incorporated by reference in the registration statement, or is contained in a form of prospectus filed pursuant to Rule 424(b) that is part of the registration statement;
b. | That, for the purpose of determining any liability under the Securities Act, each such post‑effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof; and |
c. | To remove from registration by means of a post‑effective amendment any of the securities being registered which remain unsold at the termination of the offering. |
d. | That, for the purpose of determining liability under the Securities Act to any purchaser: |
i. | Each prospectus filed by a registrant pursuant to Rule 424(b)(3) shall be deemed to be part of the registration statement as of the date the filed prospectus was deemed part of and included in the registration statement; and |
II-2
ii. | Each prospectus required to be filed pursuant to Rule 424(b)(2), (b)(5) or (b)(7) as part of a registration statement in reliance on Rule 430B relating to an offering made pursuant to Rule 415(a)(1)(i), (vii) or (x) for the purpose of providing the information required by Section 10(a) of the Securities Act shall be deemed to be part of and included in the registration statement as of the earlier of the date such form of prospectus is first used after effectiveness or the date of the first contract of sale of securities in the offering described in the prospectus. As provided in Rule 430B, for liability purposes of the issuer and any person that is at that date an underwriter, such date shall be deemed to be a new effective date of the registration statement relating to the securities in the registration statement to which the prospectus relates, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such effective date, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such effective date. |
e. | That, for the purpose of determining liability of the registrant under the Securities Act to any purchaser in the initial distribution of the securities, the undersigned registrant hereby undertakes that in a primary offering of securities of the undersigned registrant pursuant to this registration statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser: |
i. | Any preliminary prospectus or prospectus of the undersigned registrant relating to the offering required to be filed pursuant to Rule 424; |
ii. | Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned registrant or used or referred to by the undersigned registrant; |
iii. | The portion of any other free writing prospectus relating to the offering containing material information about the undersigned registrant or its securities provided by or on behalf of the undersigned registrant; and |
iv. | Any other communication that is an offer in the offering made by the undersigned registrant to the purchaser. |
(2) | The undersigned registrant hereby undertakes that, for purposes of determining any liability under the Securities Act, each filing of the registrant’s annual report pursuant to Section 13(a) or Section 15(d) of the Exchange Act (and, where applicable, each filing of an employee benefit plan’s annual report pursuant to Section 15(d) of the Exchange Act) that is incorporated by reference in the registration statement shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
(3) | The undersigned registrant hereby undertakes that: |
a. | For purposes of determining any liability under the Securities Act, the information omitted from the form of prospectus filed as part of this registration statement in reliance on Rule 430A and contained in a form of prospectus filed by the undersigned registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective; and |
b. | For the purpose of determining any liability under the Securities Act, each post‑effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
II-3
(4) | The undersigned registrant hereby undertakes to file an application for the purpose of determining the eligibility of the trustee to act under subsection (a) of Section 310 of the Trust Indenture Act of 1939 (the “Trust Indenture Act”) in accordance with the rules and regulations prescribed by the SEC under Section 305(b)(2) of the Trust Indenture Act. |
(5) | Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the undersigned registrant, pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the undersigned registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue. |
II-4
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant certifies that it has reasonable grounds to believe that it meets all the requirements for filing on Form S‑3 and has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in Boulder, Colorado, on February 10, 2020.
BRICKELL BIOTECH, INC. | ||
By: | /s/ Robert B. Brown | |
Robert B. Brown Chief Executive Officer | ||
SIGNATURES AND POWER OF ATTORNEY
KNOW ALL PERSONS BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Robert B. Brown and R. Michael Carruthers, and each of them, as his or her true and lawful attorney‑in‑fact and agent, with full power of substitution and resubstitution, for him or her and in his or her name, place and stead, in any and all capacities, to sign any and all amendments to this registration statement, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys‑in‑fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he or she might or could do in person, hereby ratifying and confirming all that said attorneys‑in‑fact and agents, or any of them or their or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons on February 10, 2020, in the capacities indicated.
Signature | Title |
/s/ ROBERT B. BROWN | Chief Executive Officer and Director (Principal Executive Officer) |
Robert B. Brown | |
/s/ R. MICHAEL CARRUTHERS | Chief Financial Officer (Principal Financial Officer) |
R. Michael Carruthers | |
/s/ JOSE BRETON | Controller and Chief Accounting Officer (Principal Accounting Officer) |
Jose Breton | |
/s/ REGINALD L. HARDY | Co-Founder and Chairman of the Board of Directors |
Reginald L. Hardy | |
/s/ GEORGE ABERCROMBIE | Director |
George Abercrombie | |
/s/ DENNISON T. VERU | Director |
Dennison T. Veru | |
/s/ VIJAY B. SAMANT | Director |
Vijay B. Samant | |
/s/ GARY A. LYONS | Director |
Gary A. Lyons | |
II-5
EXHIBIT INDEX
Exhibit Number | Description of Exhibit | |
1.1+ | Form of Underwriting Agreement. | |
Restated Certificate of Incorporation, as currently in effect (incorporated by reference to Exhibit 3.2 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | ||
Amended and Restated Bylaws (incorporated by reference to Exhibit 3.3 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | ||
Specimen Common Stock Certificate (incorporated by reference to Exhibit 4.1 to the Company’s registration statement on Form S-8 filed with the SEC on September 10, 2019). | ||
Form of Indenture. | ||
4.3+ | Form of Warrant Agreement and Warrant Certificate. | |
4.4+ | Form of Unit Agreement. | |
Opinion of Mayer Brown LLP. | ||
10.1† | License, Development and Commercialization Agreement, as amended, dated March 31, 2015, by and between Brickell Biotech, Inc. and Kaken Pharmaceutical Co., Ltd. (incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | |
10.2† | Right of First Negotiation Agreement, as amended, dated March 31, 2015, by and between Brickell Biotech, Inc. and Kaken Pharmaceutical Co., Ltd. (incorporated by reference to Exhibit 10.3 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | |
10.3† | License Agreement, as amended, dated December 15, 2012, by and among Brickell Biotech, Inc., Bodor Laboratories, Inc. and Nicholas S. Bodor (incorporated by reference to Exhibit 10.4 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | |
10.4† | UAB Research Foundation License Agreement, as amended, dated June 26, 2012, by and between Brickell Biotech, Inc. and the UAB Research Foundation (incorporated by reference to Exhibit 10.5 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | |
10.5† | License Agreement, dated May 20, 2011, by and between Brickell Biotech, Inc. and the University of Manchester (incorporated by reference to Exhibit 10.6 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | |
10.6† | License Agreement, as amended, dated June 6, 2013, by and among Brickell Biotech, Inc, Orca Pharmaceuticals LLC and the New York University (incorporated by reference to Exhibit 10.7 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | |
10.7† | Orca Pharmaceuticals LLC Asset Purchase Agreement, dated November 23, 2015 by and between Brickell Biotech, Inc. and Orca Pharmaceutics (incorporated by reference to Exhibit 10.8 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | |
10.8† | Panmira Pharmaceuticals LLC Purchase Agreement, dated January 30, 2015, by and between Brickell Biotech, Inc. and Panmira Pharmaceuticals (incorporated by reference to Exhibit 10.9 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | |
Boulder Lease Agreement, as amended, dated August 4, 2016, by and between Brickell Biotech, Inc. and BMC Properties, LLC (incorporated by reference to Exhibit 10.10 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | ||
Employment Agreement, dated November 16, 2018, by and between Brickell Biotech, Inc. and Robert Brown (incorporated by reference to Exhibit 10.11 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | ||
Second Amended and Restated Employment Agreement, dated November 27, 2018, by and between Brickell Biotech, Inc. and Andy Sklawer (incorporated by reference to Exhibit 10.12 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | ||
Employment Agreement, dated August 1, 2016, and Amendment to Employment Agreement, dated August 28, 2019, by and between Brickell Biotech, Inc. and Deepak Chadha (incorporated by reference to Exhibit 10.13 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | ||
Brickell Biotech, Inc. Letter Agreement, dated July 10, 2018 by and between Brickell Biotech Inc. and Jose Breton (incorporated by reference to Exhibit 10.14 to the Company’s Current Report on Form 8-K filed with the, SEC on September 3, 2019). | ||
Employment Agreement, dated July 1, 2019, and Amendment to Employment Agreement, dated August 27, 2019, by and between Brickell Biotech, Inc. and David R. McAvoy (incorporated by reference to Exhibit 10.15 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | ||
II-6
Employment Agreement, dated August 1, 2019, by and between Brickell Biotech, Inc. and Adam Levy (incorporated by reference to Exhibit 10.16 to the Company’s Current Report on Form 8-K filed with the SEC on September 3, 2019). | ||
Employment Agreement, dated October 3, 2019, by and between Brickell Biotech, Inc. and Dr. Sanjeev Ahuja (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed with the SEC on October 9, 2019). | ||
Settlement and Termination Agreement, dated November 25, 2019, by and between Brickell Subsidiary, Inc., Brickell Biotech, Inc. and NovaQuest Co-Investment Fund X, L.P. (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K filed with the SEC on November 25, 2019). | ||
Consent of Ernst & Young LLP. | ||
Consent of Mayer Brown LLP (included as part of Exhibit 5.1). | ||
Power of Attorney (included in the signature page to this registration statement). | ||
25.1* | Statement of Eligibility of Trustee under the Indenture. | |
* | To be filed separately in accordance with Section 305(b)(2) of the Trust Indenture Act of 1939, as amended, and the appropriate rules and regulations thereunder. |
+ | To be filed, if applicable, by amendment or as an exhibit to a document to be incorporated by reference or deemed to be incorporated by reference in this registration statement, including a Current Report on Form 8-K. |
† | Certain confidential information contained in this agreement has been omitted because it (i) is not material and (ii) would be competitively harmful if publicly disclosed. |
II-7